
Abstract
Purpose
Alterations in the urinary microbiome have been associated with urological diseases. The microbiome of patients with urethral stricture disease (USD) remains unknown. Our objective is to examine the microbiome of USD with a focus on inflammatory USD caused by lichen sclerosus (LS).
Methods
We collected mid-stream urine samples from men with LS-USD (cases; n = 22) and non-LS USD (controls; n = 76). DNA extraction, PCR amplification of the V4 hypervariable region of the 16S rRNA gene, and sequencing was done on the samples. Operational taxonomic units (OTUs) were defined using a > 97% sequence similarity threshold. Alpha diversity measurements of diversity, including microbiome richness (number of different OTUs) and evenness (distribution of OTUs) were calculated and compared. Microbiome beta diversity (difference between microbial communities) relationships with cases and controls were also assessed.
Results
Fifty specimens (13 cases and 37 controls) produced a 16S rRNA amplicon. Mean sample richness was 25.9 vs. 16.8 (p = 0.076) for LS-USD vs. non-LS USD, respectively. LS-USD had a unique profile of bacteria by taxonomic order including Bacillales, Bacteroidales and Pasteurellales enriched urine. The beta variation of observed bacterial communities was best explained by the richness.
Conclusions
Men with LS-USD may have a unique microbiologic richness, specifically inclusive of Bacillales, Bacteroidales and Pasteurellales enriched urine compared to those with non-LS USD. Further work will be required to elucidate the clinical relevance of these variations in the urinary microbiome.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Mounting evidence suggests that the urinary tract harbors a unique microbiome, which encompasses the microorganisms in a particular environment and their combined genetic material and may have an impact on lower urinary tract function [1,2,2]. A unique microbiome in women has recently been discovered [3], deepening our comprehension of common lower urinary tract ailments, including urinary incontinence and urinary tract infection [4]. Likewise, the male urethra has been shown to harbor varied bacteria [5, 6]. The study of the male urinary microbiome remains in its infancy and relationships between lower urinary tract symptoms and variance in microbiota are not well established [2].
Alterations of the typical microbiome have been implicated in chronic low-level inflammation throughout the body [7]. For example, perturbation to the composition or function of the gut microbiome is associated with a wide range of disease such as arthritis, frailty, metabolic disease, neurologic disease, and inflammatory bowel conditions [8]. More recent studies have offered mechanistic insights into how altered microbiomes promote disease, highlighting microbial production of bioactive small molecules as a relatively underappreciated mechanism by which microbiomes elicit their effect on host immunity [9]. The degree to which alterations in the urinary microbiome affect the male urinary tract or contribute to chronic genitourinary inflammation, however, is unknown.
One poorly understood inflammatory condition that negatively impacts the male urethra is genitourinary lichen sclerosus (LS) [10]. LS is a chronic, inflammatory condition of unknown etiology that can result in urethral stricture disease (USD) [11]. USD incurs substantial morbidity and incurs high costs in affected men [12]. Infectious etiologies have been suggested as causative agents for LS [13]. Explorations of the urinary microbiome may shed light into the pathophysiology of this complex disease.
Our objective was to determine the typical microbiota in patients with USD. Second, we aim to define whether associations exist between microbiota and a special population of USD patients: those with LS. We hypothesize differences exist in microbial diversity and richness between patients with LS-associated USD compared to those with non-LS-associated USD.
Methods
Patient population
Eligible patients were identified upon their presentation in clinic and prospectively recruited for inclusion without incentive. Inclusion criteria included men age > 18 years old, with suspected LS-USD (cases; n = 22) or non-LS USD (controls; n = 76) presenting for diagnosis and work-up. Patients were excluded if they had recent genitourinary surgery/instrumentation (< 2 months prior to study sample collection), a recent symptomatic urinary tract infection (< 6 months) or took recent antibiotics (< 2 months). Patients were also excluded if they were HIV positive, and/or on immunosuppressant or systemic steroid therapy, and/or had a systemic autoimmune disease (such as lupus, multiple sclerosis, glomerulonephritis, Graves’ disease, or rheumatoid arthritis). All patients underwent traditional urine culture as well as simultaneous urinary microbiome analysis. This study was approved by University of California, San Francisco Internal Review Board.
For those meeting inclusion criteria, we collected a mid-stream urine sample at time of the office visit, at least 30 days before the procedure, prior to administration of any peri-procedural antibiotics or diagnostic studies. Patients with urine cultures resulting in > 100,000 colony forming units were later excluded. Men were characterized by presenting phenotype as LS-USD (cases) or non-LS USD (controls). A diagnosis of LS was suspected clinically based on physical exam findings by reconstructive expert (BB) and included: white skin changes on the glans, meatus, fusion of glans to foreskin, and/or LS urethral changes such as stippled or pan-urethral disease on retrograde urethrogram. A diagnosis of USD was suspected by patient reported symptoms of poor urinary flow, referral physician suspected diagnosis and confirmed by retrograde urethrogram. All LS patients included in this study, at the minimum, had meatal stenosis. Surgical treatment for USD or LS was not necessary for inclusion.
Urinary microbiome analysis
Urine samples underwent DNA extraction using an optimized Cetyl trimethylammonium bromide (CTAB) method followed by PCR amplification of the V4 hypervariable region of the 16S rRNA gene, and sequencing on the NextSeq platform (Illumina, San Diego, California, USA) as previously described [14]. This was triplicated. Paired sequencing reads were quality filtered and clustered into operational taxonomic units (OTUs) using a 97% sequence similarity threshold while removing chimeras using the cluster_otus command in usearch (v8.0.1623). Read counts for OTUs detected at < 0.001% of the total read count were discarded to minimize noise in the dataset [15]. Sequence reads were representatively rarefied to 1000 reads per sample. Alpha diversity is a metric of mean species diversity in a particular environment, whereas the differentiation among the inhabitants is beta diversity [16]. Alpha diversity (Shannon, Simpson, Inverse Simpson, and Faith’s phylogenetic diversity) indices of microbiota richness (number of different OTUs) and evenness (distribution of OTUs), were calculated using the vegan package in R. Beta diversity (Bray–Curtis dissimilarity, Canberra distance, and weighted/unweighted UniFrac analysis) distances matrices were calculated in QIIME (Python script) [17] and visualized using principal coordinate analysis.
Statistical analyses
Alpha diversity metrics were compared using Student’s or Welch’s T test where appropriate. Permutational multivariate analysis of variance (PERMANOVA) was used to determine relationships between clinical phenotype and beta diversity. Taxon relative abundance comparisons were performed using the three-model approach we have previously described, which accounts for varying data distributions and the zero-inflated nature of count-based microbiota sequence datasets [14]. DESeq was used to determine sample enrichment [17]. Patient characteristics were compared using Chi-squared, fisher’s exact or t tests as appropriate. Stata 15 (College Station, Texas, USA) and the R statistical environment were used for all statistical analysis. p values less than 0.05 were considered statistically significant.
Results
Initially 98 patients were recruited: 22 cases and 76 controls. Our population reflects our role as a tertiary referral center for the surgical management of USD with 34.7% of patients already having undergone at least one urethroplasty. Similarly, all comers with USD averaged three prior urethral dilations. Among those included, a greater proportion of LS-USD patients versus non-LS USD patients (53.9% and 24.3%, respectively) had used antibiotics within the 12 months prior to study recruitment, though this finding was not statistically significant (p = 0.08). Screening urine culture eliminated 16 patients from inclusion due to the presence of > 100,000 colony forming units on traditional urine culture. Fifty of the remaining 82 samples successfully produced a 16S rRNA amplicon for microbiota analyses. (Supplemental Fig. 1). Patients (LS-USD or non-LS USD) included in these analyses were similar with respect to age, IPSS, and procedural history (Table 1). Median BMI for the LS-USD patients was 32 [interquartile range (IQR) 28–34] vs. 27 (IQR 25–30; p value = 0.02) for non-LS USD. Of note, there were no statistically significant differences in baseline characteristics, IPSS scores, or procedural history for those patients with urine samples ultimately included vs. not included.
Patients underwent full diagnostic work-up for USD. Treatment elected by patients ultimately included in the study included: 38% excision and primary anastomosis/non-transecting urethroplasty, 24% buccal graft urethroplasty, 16% direct visual internal urethrotomy (DVIU), and 22% conservative management for their disease. Among LS-USD patients, 30.1% underwent urethroplasty, 23.0% underwent phalloplasty and panniculectomy, 23.0% underwent DVIU, and 23.0% used calibration and steroid cream alone as therapy. There was a significant difference in stricture location for LS-USD patients versus non-LS USD patients, with a predominance of bulbar strictures in the non-LS group. (Table 1).
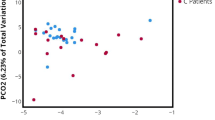
Mean microbiome richness was low and trended towards being lower in non-LS USD patients (25.9 vs. 16.8 for LS-USD vs. non-LS USD, respectively; p = 0.076, Fig. 2). Mean Faith’s phylogenetic diversity (taxon richness) also trended lower 4.23 vs. 3.05 (LS-USD and non-LS USD, respectively; p = 0.058) in patients without underlying LS (Fig. 1). Though not statistically significant based on predetermined cutoffs, these data suggest that LS-USD is associated with the presence of a greater number of types of bacterial in urine samples.

Between-group conparison of alpha diversity indices indicate that LS-USD patient urine harbors a greater number of bacterial types and greater diversity compared with non-LS USD patients
To assess whether relationships existed between urine microbiota and clinical phenotype, unweighted and weighted UniFrac distance matrices (metric for comparing biological communities) were constructed and analyzed. We did not find a significant relationship between LS-USD and non-LS USD patient microbiota (Supplemental Table 1). In contrast, microbiota richness was strongly and significantly related to urine microbiota suggesting that the observed variation in community structure was primarily driven by the number of types of bacteria detected rather than the presence or absence of LS.
Since it is likely that the majority of organisms detected in the urine of USD patients are common to both LS- and non-LS patients, and that LS may be associated with expansion of a small number of specific bacteria, we next performed comparison of organisms detected between LS- and non-LS patients. Patients with LS-USD exhibited enrichment of Sneathia, Lactobacillus, and Tissierellaceae while Staphylococcus, Facklamia, and Dialister were enriched in urine samples of patients with non-LS USD (Figs. 2, 3).

Microbiome diversity to order taxonomy level, by sample

Microbiome diversity to order taxonomy level, averaged by group
Discussion
To our knowledge, this is the first study of the urinary microbiome in patients with USD and specifically, LS. In the USD population, urine was enriched with unique bacterial communities. Based on our data, the microbiota of LS-USD patients appears to house greater bacterial diversity compared with patients with non-LS USD. Of note, the LS-USD group had twice the rate of antibiotic exposure than non-LS USD patients, and increased median BMI. Strictures associated with genitourinary LS were noted in the meatus, penile urethra, and a pan-urethral manner vs. our control population wherein bulbar strictures predominated. Though the variance in overall microbiota composition was not related to clinical phenotype, specific bacteria genera were found to be enriched in the urine of patients with LS-USD. These included Sneathia and Lactobacillus taxa. While Lactobacillus represents the dominant genus in the female vaginal tract, certain Lactobacillis species have been associated with urge incontinence [18]. A unique microbiologic signature of LS could lead to the development of non-invasive testing for LS, help to explain pathophysiology of the disease, or lend itself to targeted intervention. Prior studies in healthy men have demonstrated an abundance of Enterobacteriales, specifically Enterobacteriaceae in urine samples [19]. A wide range of microbiome composition has been reported in small studies to include: Sneathia, Veillonella, Corynebacterium, Prevotella, Streptococcus and Ureaplasma, Corynebacterium, Staphylococcus, and Streptococcus [6, 20]. We note important differences in median BMI between LS-USD and non-LS USD groups [21]. There is no established normal microbiota signature in the male urinary tract stratified by patient BMI with research in this area still in its infancy.
The potential clinical and research implications of increased microbiota richness of LS are intriguing. LS is associated with squamous cell carcinoma and a suitable biomarker to aid in identification of malignant potential would be of high utility [22]. Alterations to a cell’s microenvironment by local bacteria may explain the pathway between inflammation and cancer development [23]. Efforts to identify unique protein expression profiles of LS are underway, but the microbiome itself may be a potential biomarker [13, 24]. Given the complexity and potential morbidity in obtaining a tissue diagnosis of LS in the urethra, using urine as a microbial-based biomarker would be advantageous.
While an infectious etiology of LS has been actively studied, there is no consensus that bacterial infection is a cause [10]. While some groups have implicated Borrelia burgdorferi [25], evidence is ultimately poor with an unknown direction of association. In our cohort, we did not find evidence of this bacterium for patients in the LS-USD group. There is alternative evidence of a viral cause of LS [13]. Additionally, how microbiota may inform patients’ USD symptomatology or affect surgical repair is an open question. In abdominal surgery, for example, the microbiome may complicate anastomotic healing [26]. In this series, we did not correlate microbiological findings with surgical outcomes.
While our exclusion criteria precluded recent procedure or UTI, our patients had frequent prior endoscopic interventions in their past. These procedures may reflect higher baseline level of bacterial colonization and may explain differences in lower urinary tract microbiota than those found previously [5, 19, 20]. Alternatively, prior procedures may indicate higher rates of remote antibiotic exposures and atypical bacterial populations [27]. Patients considering surgical and non-surgical LUTS treatment had detectable bacteria in specimens 98% of the time, suggesting bacteria may be present in the urinary tract independent of prior procedure [2]. Regardless of sexually transmitted infection status, Dong et al. detected an average of 18 bacterial genera among a group of 32 healthy young men [6]. The microbiota of the male urinary tract is certainly more complex and nuanced than traditional pre-operative urine cultures would suggest [28].
Limitations
LS diagnosis was based on clinical exam findings at their initial visit alone and not confirmed by pathologic diagnosis. Only 60% of samples underwent successful amplification, but there were no differences in demographics between those samples that were successful or not so we do not expect this would bias our results. This degree of sample loss is also in line with prior literature [29, 30]. Our small sample size leaves us unable to identify significant differences in specific bacterial taxa based on stricture etiology. The effect of steroid use (present for 23% of the cohort) has unknown impact on the urinary microbiome. This work does not allow us to determine causality given the lack of longitudinal data. Increased richness in the LS cohort may imply less chance of predominance by single organisms; however, the number of LS patients in our cohort is quite small (n = 13). While LS can present primarily as dermatological condition, our patients were referred for urethral stricture disease and we did not pursue glans swabs in the study. It is unknown if patients with primary skin changes on the glans or penile shaft skin without USD would have detectable differences within the urinary microbiome. A small prior study suggested higher detectable rates of bacteria using catheterized specimens. Catheterization was not viable given USD and we elected to pursue voided samples to aid recruitment [2]. In addition, we did not utilize controls for comparison to rule out contamination. In addition, the collection of urine mid-stream may represent the urethral microbiome. The inclusion criteria for microbiome studies based on the method of urine collection or antibiotic washout period is not standardized [29]. Patients with standard urine cultures > 100,000 cfu/ml were excluded, which may lead to bias in the results. Common to patients with USD, ours reported multiple prior instrumentations, infections, and/or dilation with catheterization which could confound results by altering native urinary microbial communities. Finally, the microbiome may reflect the local and regional environment of our hospital or potentially even patients’ immediate environment or close relationships [31, 32].
Despite these limitations, we present the first study on the microbiome of patients with USD. Further research on the unique microbiome of these patients may lead to earlier detection of pathology and a unique avenue of studying the pathophysiology of this disease. In particular, this represents a novel approach to understand LS-USD.
Conclusion
We identify a discriminatory bacterial signature in the urine of patients with LS-USD vs. non-LS USD which requires validation in future studies. The implications of the microbiome in genitourinary pathology are evolving and further research is needed.
References
Drake MJ, Morris N, Apostolidis A et al (2017) The urinary microbiome and its contribution to lower urinary tract symptoms; ICI-RS 2015. Neurourol Urodyn 36:850–853. https://doi.org/10.1002/nau.23006
Bajic P, Van Kuiken ME, Burge BK et al (2018) Male Bladder Microbiome Relates to Lower Urinary Tract Symptoms. European Urology Focus. https://doi.org/10.1016/j.euf.2018.08.001
Mueller ER, Wolfe AJ, Brubaker L (2017) Female urinary microbiota. Curr Opin Urol 27(3):282–286
Schneeweiss J, Koch M, Umek W (2016) The human urinary microbiome and how it relates to urogynecology. Int Urogynecol J 27:1307–1312. https://doi.org/10.1007/s00192-016-2944-5
Frølund M, Wikström A, Lidbrink P et al (2018) The bacterial microbiota in first-void urine from men with and without idiopathic urethritis. PLoS ONE 13:e0201380. https://doi.org/10.1371/journal.pone.0201380
Dong Q, Nelson DE, Toh E et al (2011) The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS ONE 6:e19709. https://doi.org/10.1371/journal.pone.0019709
Belkaid Y, Hand TW (2014) Role of the microbiota in immunity and inflammation. Cell 157:121–141. https://doi.org/10.1016/j.cell.2014.03.011
Buford TW (2017) (Dis)Trust your gut: the gut microbiome in age-related inflammation, health, and disease. Microbiome 5:80. https://doi.org/10.1186/s40168-017-0296-0
Fischbach MA, Segre JA (2016) Signaling in host-associated microbial communities. Cell 164:1288–1300. https://doi.org/10.1016/j.cell.2016.02.037
Grimes MD, Tesdahl BA, Schubbe M et al (2019) Histopathology of anterior urethral strictures: towards a better understanding of stricture pathophysiology. J Urol. https://doi.org/10.1097/JU.0000000000000340
Pugliese JM, Morey AF, Peterson AC (2007) Lichen sclerosus: review of the literature and current recommendations for management. J Urol 178:2268–2276. https://doi.org/10.1016/j.juro.2007.08.024
Osterberg EC, Murphy G, Harris CR et al (2017) Cost-effective strategies for the management and treatment of urethral stricture disease. Urol Clin N Am 44:11–17. https://doi.org/10.1016/j.ucl.2016.08.002
Levy A, Browne B, Fredrick A et al (2019) Insights into the pathophysiology of urethral stricture disease due to lichen sclerosus: comparison of pathological markers in lichen sclerosus induced strictures vs nonlichen sclerosus induced strictures. J Urol 201:1158–1163. https://doi.org/10.1097/JU.0000000000000155
Fujimura KE, Sitarik AR, Havstad S et al (2016) Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med 22:1187–1191. https://doi.org/10.1038/nm.4176
Whittaker RH (1972) Evolution and measurement of species diversity. Taxon 21:213. https://doi.org/10.2307/1218190
Caporaso JG, Kuczynski J, Stombaugh J et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. https://doi.org/10.1186/s13059-014-0550-8
Pearce MM, Hilt EE, Rosenfeld AB et al (2014) The female urinary microbiome: a comparison of women with and without urgency urinary incontinence. Blaser MJ, ed. MBio. https://doi.org/10.1128/mBio.01283-14
Wu P, Zhang G, Zhao J et al (2018) Profiling the urinary microbiota in male patients with bladder cancer in China. Front Cell Infect Microbiol 8:167. https://doi.org/10.3389/fcimb.2018.00167
Shrestha E, White JR, Yu S-H et al (2018) Profiling the urinary microbiome in men with positive versus negative biopsies for prostate cancer. J Urol 199:161–171. https://doi.org/10.1016/j.juro.2017.08.001
Gérard P (2016) Gut microbiota and obesity. Cell Mol Life Sci 73:147–162. https://doi.org/10.1007/s00018-015-2061-5
Carlson BC, Hofer MD, Ballek N et al (2013) Protein markers of malignant potential in penile and vulvar lichen sclerosus. J Urol 190:399–406. https://doi.org/10.1016/j.juro.2013.01.102
Sfanos KS, Yegnasubramanian S, Nelson WG et al (2018) The inflammatory microenvironment and microbiome in prostate cancer development. Nat Rev Urol 15:11–24. https://doi.org/10.1038/nrurol.2017.167
Rajpoot M, Sharma AK, Sharma A et al (2018) Understanding the microbiome: emerging biomarkers for exploiting the microbiota for personalized medicine against cancer. Semin Cancer Biol 52:1–8. https://doi.org/10.1016/j.semcancer.2018.02.003
Bunker CB, Shim TN (2015) Male genital lichen sclerosus. Indian J Dermatol 60(2):111–117
Shogan BD, Smith DP, Christley S et al (2014) Intestinal anastomotic injury alters spatially defined microbiome composition and function. Microbiome 2:35. https://doi.org/10.1186/2049-2618-2-35
Kassiri B, Shrestha E, Kasprenski M et al (2019) A prospective study of the urinary and gastrointestinal microbiome in prepubertal males. Urology. https://doi.org/10.1016/j.urology.2019.05.031
Sathiananthamoorthy S, Malone-Lee J, Gill K et al (2018) Reassessment of routine midstream culture in diagnosis of urinary tract infection. Munson E, ed. J Clin Microbiol. https://doi.org/10.1128/JCM.01452-18
Pollock J, Glendinning L, Wisedchanwet T et al (2018) The madness of microbiome: attempting to find consensus “best practice” for 16S microbiome studies. Liu S-J, ed. Appl Environ Microbiol. https://doi.org/10.1128/AEM.02627-17
Brooks JP, Edwards DJ, Harwich MD et al (2015) The truth about metagenomics: quantifying and counteracting bias in 16S rRNA studies. BMC Microbiol 15:66. https://doi.org/10.1186/s12866-015-0351-6
Dill-McFarland KA, Tang Z-Z, Kemis JH et al (2019) Close social relationships correlate with human gut microbiota composition. Sci Rep 9:703. https://doi.org/10.1038/s41598-018-37298-9
Lax S, Sangwan N, Smith D et al (2017) Bacterial colonization and succession in a newly opened hospital. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aah6500
Funding
This work was made possible by generous gifts from Jonathan Kaplan, the Alafi fund, and Anita and Kevan Del Grande Funding was provided by Alafi Foundation (Grant no. 10001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Cohen, A.J., Gaither, T.W., Srirangapatanam, S. et al. Synchronous genitourinary lichen sclerosus signals a distinct urinary microbiome profile in men with urethral stricture disease. World J Urol 39, 605–611 (2021). https://doi.org/10.1007/s00345-020-03198-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00345-020-03198-9