
Abstract
Immunotherapy is rapidly changing the field of urologic oncology. In this review, we discuss the role of the immune system in general and place a particular emphasis on the biology of the immune checkpoint and its role in cancer. Bladder cancer, as one of the most immunogenic neoplasms, is an exciting target for immune checkpoint inhibition. Early preclinical data and human trial experience suggest that this new drug class may shape bladder cancer therapy for years to come.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immunotherapy and immune checkpoint inhibition in particular present an exciting opportunity for the treatment of bladder cancer. Over the last 30 years, bladder cancer patients have seen few advances in the treatment of their disease. With an estimated 74,000 new cases and 16,000 deaths from bladder cancer in 2015, the incidence and survival have remained relatively constant [1, 2]. In patients, both with muscle-invasive disease undergoing radical cystectomy as well as those with locally advanced or metastatic disease, there have been no new FDA-approved therapies for those who cannot tolerate or fail to respond to cisplatin-based chemotherapy. However, in the last several years, new insights into tumor immunology have lead to the development of a new class of drugs termed immune checkpoint inhibitors, several of which have demonstrated impressive anti-tumor responses in several malignancies, including melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma (RCC) [3–8].
Currently, these immune checkpoint inhibitors are being actively studied in several treatment settings for bladder cancer, including for non-muscle-invasive disease with BCG (pembrolizumab, NCT02324582) as well as neoadjuvant or adjuvant therapy after cystectomy (atezolizumab, NCT02451423, NCT02450331). In June 2014, the FDA granted the anti-PD-L1 antibody atezolizumab (MPDL3280A) “breakthrough” status for urothelial carcinoma based on promising results of a phase 1a trial in patients with metastatic disease [9].
The purpose of this article is to review the basis for immune checkpoint inhibition in muscle-invasive bladder cancer and discuss the current state of clinical trials to evaluate their safety and efficacy.
Cancer immunotherapy and the role of the immune checkpoint
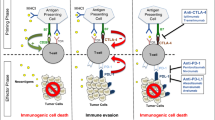
Human tumors elicit adaptive immune responses, mediated primarily by T lymphocytes. T cells have been the primary focus of cancer immunotherapy primarily due to their ability to organize diverse immune responses via CD4+ helper T cells that have adaptive and innate effector mechanisms. Analysis of immune infiltrates suggests that greater infiltration by T lymphocytes is largely associated with a stronger anti-tumor activity and chemotherapeutic response [10–12]. Broad cytotoxic CD8+ T cell infiltration in particular has been associated with improved survival through its role of recognizing tumor-associated antigens (TAA) presented by major histocompatibility complex class I (MHC-I) molecules [13]. CD4+ T cells also exhibit effector functions against MHC class II molecule-negative tumors and produce cytokines that mediate these immune responses. These effector T cells are balanced by Foxp3+ regulatory Treg (T) cells, which suppress natural killer cells and the innate immune response as well as effector T cells and the adaptive response [14]. The balance of co-stimulatory and inhibitory responses to cancer is a central tenant of cancer immunology.
Tumors evade the immune system primarily via (1) decreasing MHC-I expression, and in turn decrease CD8+ activity; (2) defective antigen processing and presentation which causes decreased recognition by T cells; and (3) increased expression of co-inhibitory (i.e., immune checkpoint) molecules. This final mechanism of immune evasion is the focus of this review.
In a non-tumor environment, immune checkpoints are crucial to regulate the immune system and prevent autoimmunity. The most studied and clinically relevant checkpoint proteins are programmed cell death (PD)-1, PD-ligand-1 (PD-L1), and cytotoxic T lymphocyte-associated protein 4 (CTLA-4). Immune checkpoint expression can be dysregulated by tumors, and the current role of cancer immunotherapy seeks to restore T cell-mediated immune response [15].
It is in this context that monoclonal antibodies to PD-1 (nivolumab), PD-L1 (atezolizumab), and CTLA-4 (ipilimumab) have been formulated. These immune checkpoint inhibitors restore the effector T cell anti-tumor activity primarily by blocking the immune checkpoint’s normal signal to stop a cell’s immune response.
The CTLA-4 checkpoint
CTLA-4, which is expressed solely on T cells, primarily inactivates T cell activity by competing with the CD28 co-stimulatory molecule [16]. CD28 and CTLA-4 share the identical ligands of CD80 and CD86 on antigen-presenting cells (APCs), and thus CTLA-4 competes with CD28 function in T cell survival, proliferation, and recruitment [17, 18]. In particular, CTLA-4 down-modulates CD4+ helper T cell activity and enhances Treg immunosuppressive functions [19].
The blockade of CTLA-4 has been in development for sometime, since Allison and colleagues used preclinical models to show that antibody blockade of CTLA-4-enhanced immune-mediated anti-tumor activity [20]. Ipilimumab is a monoclonal antibody targeting CTLA-4 and the first therapy to demonstrate a survival benefit for patients with metastatic melanoma, and it was quickly FDA-approved thereafter (see Fig. 1) [7]. More impressive was that 18 % of patients survived beyond 2 years, compared with a 5 % survival rate with the previous standard of care. However, the potent immunomodulatory effects of CTLA-4 blockade leads to a significant adverse events (AE), which occur in >70 % of patients treated with ipilimumab [21]. These range from dermatitis, colitis, and hepatitis, to less common uveitis, neuropathy, and lupus nephritis [22]. Essentially with anti-tumor immune suppression comes a component of autoimmune suppression [23].

Abridged timeline of immune checkpoint drug approval
PD-1 checkpoint
It is in the context of CTLA-4’s dramatic anti-tumor activity with a high burden of AEs that propagated interest in the PD-1 pathway. In contrast to CTLA-4, PD-1 expression is induced in peripheral tissues when T cells become activated. This cell-surface molecule is activated by two ligands—PD-L1 and PD-L2, which share 37 % sequence homology and lie within 100 kb of one another in the genome [15]. PD-1 is expressed on many different subtypes of tumor infiltrating leukocytes, and is particularly overexpressed on intra-tumoral Tregs. Similarly, PD-L1 has been shown to have high expression in several solid organ tumors, including melanoma and lung cancer [24]. PD-L2, by contrast, has been less frequently studied but is expressed on different types of APCs (monocytes, macrophages, and dendritic cells) and is also up-regulated during T cell activation in tumor [25].
PD-L1 and PD-L2 expressions can be up-regulated innately via constitutive oncogenic signaling by the tumor cells (via activation of the AKT and STAT3 pathways), or can be induced by an adaptive means as a response to inflammatory signaling [26]. Sustained ligand expression of PD-L1 or PD-L2 on tumor cells leads to proliferation of Tregs and to a state of exhaustion and ultimately T cell anergy and apoptosis. The result is an immunosuppressive state that leads to tumor cell escape and proliferation [27]. Thus far, monoclonal antibodies targeting both PD-1 (nivolumab/pembrolizumab) and PD-L1 (atezolizumab) have been evaluated in human trials. Across multiple histologies, PD-1 and PD-L1 inhibitors have shown tumor regressions and partial and complete responses [3, 5]. In some settings, response was durable beyond 2 years and persisted after drug discontinuation [28].
Rationale for use of immune checkpoint inhibition in bladder cancer
Two primary arguments for the utility of checkpoint blockade in the treatment of urothelial carcinoma are (1) new data demonstrating the high immunogenicity of bladder cancer in relation to other neoplasms and (2) the preexisting successful experience with immunotherapy for this disease.
The “immunogenicity” argument for the use of checkpoint blockade centers on the concept that a given cancer’s ability to elicit an immune response is dependent on the mutational burden of that tumor. The more mutations a tumor has, the more neoantigens are produced and presented as “non-self” to circulating T cells triggering an immune response [29].
Recently, bladder cancer has been identified as having some of the highest number of somatic mutations of any malignancy. The other cancers with high mutational burdens—melanoma and lung cancer, also occur in the setting of chronic carcinogen exposure and result in a complex interplay of many molecular errors leading to a plethora of pathways for dysplasia [30]. The relationship between mutational burden, immunogenicity, and potential immune checkpoint response also extends to individual tumors. Recent whole-exome sequencing of patients with non-small cell lung cancer (NSCLC) has shown that tumors with a higher mutational burden are more likely to respond to pembrolizumab, a PD-1 inhibitor [31].
The concept of frequent mutations causing neoantigen production and T cell recruitment and infiltration is a primary reason why immune checkpoint blockade may be successful in treating bladder cancer. T cells, particularly CD8, have been shown to predict survival in patients with muscle-invasive bladder cancer (MIBC) [32]. Additionally, overexpression of PD-L1 has been associated with both increased risk of tumor recurrence, advanced disease, and worse survival among patients with urothelial carcinoma [33, 34]. However, more recent additional studies in populations of bladder cancer patients undergoing radical cystectomy have been equivocal [35, 36].
Additionally, intravesical BCG exemplifies a 40-year precedent for using immunotherapy to treat bladder cancer. The notion that mycobacteria could be utilized as a cancer therapy was postulated by Raymond Pearl, who discovered less cancer among patients with active tuberculosis lesions [37]. It was first evaluated in humans in 1976, and since then it has become the standard first-line treatment for most forms of non-muscle-invasive bladder cancer (NMIBC) [38–40]. BCG is known to cause widespread immune activation, with T cells—particularly CD4 helper T cells, in the bladder wall after therapy, as well as a host of cytokines [41–45]. Biot et al. [46] has previously demonstrated that T cells are primed in response to BCG therapy, and these BCG-specific T cells enhance the anti-tumor immune response. Granulocytes, macrophages, and natural killer cells have all been shown to play a role in the efficacy of intravesical BCG in inducing a cytotoxic response against bladder cancer [47–49]. Though the presence of immune infiltrates after BCG is known, the exact mechanism of immune activation against tumor cells is still being evaluated.
Early studies assessing the relationship between immunotherapy response and PD-L1 expression have been mixed. Inman et al. [50] analyzed 280 patients with high-risk bladder cancer and found that PD-L1 expression was a key predictor of stage progression, with PD-L1 being most abundant in BCG-induced bladder granulomata in 11 of 12 patients failing BCG. However, more recently Hurwitz et al. [51] studied 39 patients with NMIBC and found no correlation between BCG status and PD-L1 expression. That study did suggest that PD-L1 expression increases as disease recurs, a thought-provoking finding suggesting that overexpression of the immune checkpoint and tumor escape may be a learned phenomenon.
Bladder cancer clinical trials using checkpoint inhibitors
The first human trial utilizing a checkpoint inhibitor in bladder cancer was performed by Carthon et al. [52] In that study, 12 patients with localized bladder cancer were given ipilimumab, a CTLA-4 inhibitor prior to radical cystectomy. In that study, which reported primarily grade 1–2 toxicities, an increased frequency of CD4+ ICOShi T cells were identified in the target tissue and blood. By comparing the T cell populations in the bladder cancer patient to the immune profile of a cohort of patients with metastatic melanoma, the trial was able to demonstrate that immune infiltrates can be used to predict response to checkpoint blockade and be used to guide treatment and management.
To date, the most significant data on immune checkpoint inhibition in bladder cancer have come from the phase 1a expansion trial using the anti-PD-L1 antibody atezolizumab (MPDL3280A). In the initial phase of the human trial, 67 patients with metastatic urothelial cancer were treated with anti-PD-L1 for 16 cycles or up to 1 year or if the patient developed disease progression or unacceptable toxicity. The majority of patients in the trial were smokers who had failed prior platinum-based chemotherapies. Transient elevations in IL-18, IFN-γ, and CD8+ T cells were observed during treatment. Among patients with a minimum follow-up of 6 weeks, objective response rate was 43 % (13/30) for those with strong immunohistochemistry stains for PD-L1 and 11 % (4/35) for those with weak or no PD-L1 staining. Based on this initial data, the FDA granted atezolizumab “breakthrough” status for bladder cancer [9]. In a follow-up of that study presented at ASCO in 2015, all grade treatment-related AEs were reported in 65 %, with fatigue, decreased appetite, and nausea being most common. Five percent of patients had G3-4 AEs [53]. The authors found potential biomarkers to predict response in bladder tissue, notably a myeloid gene signature including IL-1B, Cox-2, and IL-8, as well as decreased circulating inflammatory and tumor markers (CRP, HCG, CA19-9, CA-125) [54].
Additionally, pembrolizumab, an anti-PD-1 monoclonal antibody, has been studied in a cohort of patients with recurrent or metastatic urothelial cancer. Results of the phase 1b trial were recently disclosed at the 2015 ASCO meeting. In the study, 29 patients with metastatic urothelial carcinoma were evaluated, with three (10 %) complete responders and four (14 %) partial responders over duration of 15–40+ weeks. In this early disclosure, progression free survival was 8–9 weeks, and median overall survival was 9.3 months. AEs were reported in 61 % of the population, and Grade 3–4 drug-related AEs were identified in four patients (12 %), with rash being the only one seen in >1 pt. Although the data are still premature to make any definitive judgments, given that this is in a metastatic population with on average at least two prior failed therapies, the results are promising [55].
Future directions
Checkpoint inhibitors on the horizon
Although CTLA-4 and PD-1/PD-L1 are the first checkpoint inhibitors to be commercialized, they are by no means the only checkpoint targets being evaluated. There are several co-stimulatory proteins that may enhance the effects of the currently utilized checkpoint inhibitors. For example, lymphocyte activation gene3 (LAG-3) is an immune checkpoint protein highly expressed on activated T cells, as well as B cells and NK cells. T cell immunoglobulin and mucin-3 (TIM-3) is another checkpoint molecule expressed on T cells, NK cells, and monocytes. Similar to LAG-3, TIM-3 knockout mice do not develop an autoimmune phenotype, demonstrating the subtle immunomodulatory effects (as opposed to CTLA-3) of these proteins. TIM-3 and LAG-3 have both been shown to potentiate the effects of PD-1, mediated T cell response in murine models [56, 57]. Currently, an anti-Lag-3 monoclonal antibody (BMS-986016) is being evaluated in combination with nivolumab in a phase 1 trial (NCT01968109). Anti-TIM-3 antibodies have not entered human clinical trials but are being developed. Additional immune checkpoint molecules currently being evaluated as drug targets include killer inhibitory receptors (KIR), B7-H3, V-domain Ig-containing suppressor of T cell activation (VISTA), and T cell ITIM domain (TIGIT) [58].
The role of biomarkers
As mentioned, early studies of PD-1/PD-L1 inhibitors have demonstrated that pretreatment PD-L1 expression via immunohistochemistry (IHC) predicts response to PD-1 therapy. Interestingly, these analyses must occur in the target tissue, where the PD-1 pathway is thought to be most active. Conversely, patients receiving CTLA-4 undergo global T cell activation that can be measured in peripheral blood. Although PD-L1 expression by IHC predicts response, some patients without PD-L1 expression respond to therapy, while others with strong expression do not. Other potentially important biomarkers may include the expression of PD-L1 on infiltrating immune cells, the presence of CD8+ T cells in the tissue microenvironment (TME), and the presence of a deficiency in DNA mismatch repair [59–61]. Recently, Baras and colleagues evaluated expression of immune infiltrates and Pd-L1 in a group of patients with MIBC who underwent neoadjuvant chemotherapy (NAC). While no differences were found in PD-L1 expression among responders and non-responders to NAC, the ratio of CD8+ to FoxP3+ Tregs was a strong predictor of response [62]. As the quantity and diversity of immune checkpoint inhibitors increases, identifying predictive markers of response will become equally important.
Unanswered questions: the role of immune checkpoint blockade in clinical practice
Although the evidence supporting immune checkpoint blockade in bladder cancer is promising, how it should be utilized by the clinician is still an open question. Bladder cancer is treated differently according to clinical stage. For NMIBC, the use immune checkpoint blockade as an adjunct to intravesical BCG is currently being evaluated with plans for a phase 1 clinical trial (NCT02324582). However, correlative studies that describe how the immune response to BCG is modulated by checkpoint blockade are essential, both for understanding therapeutic effects as well as disease biology. Additionally, trials are underway assessing whether checkpoint blockade improves outcomes in the adjuvant (NCT02450331) and neoadjuvant (NCT02451423) settings. Lastly, in the metastatic setting, trials have thus far been performed in chemotherapy ineligible or failed populations. As biomarkers improve for therapeutic response, the potential role of immunotherapy will need to be addressed among patients unlikely to respond to cisplatin-based chemotherapy.
One area of continued interest is in the immunomodulatory effects of radiation therapy. Locally directed radiation induces DNA damage, cell cycle arrest, cell lysis, and apoptosis. This pathway has been shown to play a profound role on lymphocyte function in animal models, as the suppressive function of Tregs is impaired by the damaging effects of irradiation [63]. Radiation is also associated with increased levels of effector CD4+ and CD8+ cells and upregulates tumor-associated antigen–MHC complexes increasing their immunogenicity, which in turn enhance T cell infiltration into tumors [64]. These findings have encouraged preclinical and clinical studies combining radiation therapy and immune checkpoint blockade, and indeed this will be a major area of study in the future [65–68].
In conclusion, early data suggest that immune checkpoint blockade has a promising role in the treatment of bladder cancer. However, future research is necessary to characterize therapeutic response and identify how these drugs should be incorporated into current clinical practices.
References
Abdollah F, Gandaglia G, Thuret R, Schmitges J, Tian Z, Jeldres C, Passoni NM, Briganti A, Shariat SF, Perrotte P (2013) Incidence, survival and mortality rates of stage-specific bladder cancer in United States: a trend analysis. Cancer Epidemiol 37:219–225
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65:5–29
Brahmer JR, Tykodi SS, Chow LQ, Hwu W, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K (2012) Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med 366:2455–2465
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, Schuster SJ, Millenson MM, Cattry D, Freeman GJ (2015) PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 372:311–319
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB (2012) Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 366:2443–2454
Hamid O, Robert C, Daud A, Hodi FS, Hwu W, Kefford R, Wolchok JD, Hersey P, Joseph RW, Weber JS (2013) Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med 369:134–144
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363:711–723
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C, Lebbe C, Baurain J, Testori A, Grob J (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364:2517–2526
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, Bellmunt J, Burris HA, Petrylak DP, Teng S (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515:558–562
Fridman WH, Galon J, Pages F, Tartour E, Sautes-Fridman C, Kroemer G (2011) Prognostic and predictive impact of intra- and peritumoral immune infiltrates. Cancer Res 71:5601–5605
Pages F, Galon J, Dieu-Nosjean M, Tartour E, Sautes-Fridman C, Fridman W (2010) Immune infiltration in human tumors: a prognostic factor that should not be ignored. Oncogene 29:1093–1102
Zitvogel L, Kepp O, Kroemer G (2011) Immune parameters affecting the efficacy of chemotherapeutic regimens. Nat Rev Clin Oncol 8:151–160
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331:1565–1570
Colombo MP, Piconese S (2007) Regulatory T-cell inhibition versus depletion: the right choice in cancer immunotherapy. Nat Rev Cancer 7:880–887
Pardoll DM (2012) The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12:252–264
Schwartz RH (1992) Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 71:1065–1068
Azuma M, Ito D, Yagita H, Okumura K, Phillips JH, Lanier LL, Somoza C (1993) B70 antigen is a second ligand for CTLA-4 and CD28. Nature 366:76–79
Hathcock KS, Laszlo G, Dickler HB, Bradshaw J, Linsley P, Hodes RJ (1993) Identification of an alternative CTLA-4 ligand costimulatory for T cell activation. Science 262:905–907
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322:271–275
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736
O’Day SJ, Maio M, Chiarion-Sileni V, Gajewski TF, Pehamberger H, Bondarenko IN, Queirolo P, Lundgren L, Mikhailov S, Roman L, Verschraegen C, Humphrey R, Ibrahim R, de Pril V, Hoos A, Wolchok JD (2010) Efficacy and safety of ipilimumab monotherapy in patients with pretreated advanced melanoma: a multicenter single-arm phase II study. Ann Oncol 21:1712–1717
Fecher LA, Agarwala SS, Hodi FS, Weber JS (2013) Ipilimumab and its toxicities: a multidisciplinary approach. Oncologist 18:733–743
Gao J, He Q, Subudhi S, Aparicio A, Zurita-Saavedra A, Lee DH, Jimenez C, Suarez-Almazor M, Sharma P (2015) Review of immune-related adverse events in prostate cancer patients treated with ipilimumab: MD Anderson experience. Oncogene. doi:10.1038/onc.2015.5
Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K (2002) Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med 8:793–800
Rosenwald A, Wright G, Leroy K, Yu X, Gaulard P, Gascoyne RD, Chan WC, Zhao T, Haioun C, Greiner TC, Weisenburger DD, Lynch JC, Vose J, Armitage JO, Smeland EB, Kvaloy S, Holte H, Delabie J, Campo E, Montserrat E, Lopez-Guillermo A, Ott G, Muller-Hermelink HK, Connors JM, Braziel R, Grogan TM, Fisher RI, Miller TP, LeBlanc M, Chiorazzi M, Zhao H, Yang L, Powell J, Wilson WH, Jaffe ES, Simon R, Klausner RD, Staudt LM (2003) Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med 198:851–862
Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran Q, Lane AP, McDyer JF, Fortuno L, Schleimer RP (2005) Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol 33:280–289
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206:3015–3029
Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, Leming PD, Lipson EJ, Puzanov I, Smith DC, Taube JM, Wigginton JM, Kollia GD, Gupta A, Pardoll DM, Sosman JA, Hodi FS (2014) Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 32:1020–1030
Schumacher TN, Schreiber RD (2015) Neoantigens in cancer immunotherapy. Science 348:69–74
Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA (2013) Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214–218
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, Zappasodi R, Maeda Y, Sander C, Garon EB, Merghoub T, Wolchok JD, Schumacher TN, Chan TA (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348:124–128
Sharma P, Shen Y, Wen S, Yamada S, Jungbluth AA, Gnjatic S, Bajorin DF, Reuter VE, Herr H, Old LJ, Sato E (2007) CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma. Proc Natl Acad Sci USA 104:3967–3972
Nakanishi J, Wada Y, Matsumoto K, Azuma M, Kikuchi K, Ueda S (2007) Overexpression of B7-H1 (PD-L1) significantly associates with tumor grade and postoperative prognosis in human urothelial cancers. Cancer Immunol Immunother 56:1173–1182
Boorjian SA, Sheinin Y, Crispen PL, Farmer SA, Lohse CM, Kuntz SM, Leibovich BC, Kwon ED, Frank I (2008) T-cell coregulatory molecule expression in urothelial cell carcinoma: clinicopathologic correlations and association with survival. Clin Cancer Res 14:4800–4808
Xylinas E, Robinson B, Kluth L, Volkmer B, Hautmann R, Küfer R, Zerbib M, Kwon E, Thompson R, Boorjian S (2014) Association of T-cell co-regulatory protein expression with clinical outcomes following radical cystectomy for urothelial carcinoma of the bladder. Eur J Surg Oncol (EJSO) 40:121–127
Bellmunt J, Mullane SA, Werner L, Fay AP, Callea M, Leow JJ, Taplin ME, Choueiri TK, Hodi FS, Freeman GJ, Signoretti S (2015) Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol 26:812–817
Redelman-Sidi G, Glickman MS, Bochner BH (2014) The mechanism of action of BCG therapy for bladder cancer—a current perspective. Nat Rev Urol 11:153–162
Morales A, Eidinger D, Bruce AW (1976) Intracavitary bacillus Calmette–Guerin in the treatment of superficial bladder tumors. J Urol 116:180–183
Brausi M, Witjes JA, Lamm D, Persad R, Palou J, Colombel M, Buckley R, Soloway M, Akaza H, Böhle A (2011) A review of current guidelines and best practice recommendations for the management of nonmuscle invasive bladder cancer by the International Bladder Cancer Group. J Urol 186:2158–2167
Babjuk M, Oosterlinck W, Sylvester R, Kaasinen E, Böhle A, Palou-Redorta J, Rouprêt M (2011) EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur Urol 59:997–1008
O’Donnell MA, Luo Y, Chen X, Szilvasi A, Hunter SE, Clinton SK (1999) Role of IL-12 in the induction and potentiation of IFN-gamma in response to bacillus Calmette–Guerin. J Immunol 163:4246–4252
De Boer E, De Jong W, Steerenberg P, Aarden L, Tetteroo E, De Groot E, Van der Meijden A, Vegt P, Debruyne F, Ruitenberg E (1992) Induction of urinary interleukin-1 (IL-1), IL-2, IL-6, and tumour necrosis factor during intravesical immunotherapy with bacillus Calmette–Guerin in superficial bladder cancer. Cancer Immunol Immunother 34:306–312
Bohle A, Gerdes J, Ulmer AJ, Hofstetter AG, Flad HD (1990) Effects of local bacillus Calmette–Guerin therapy in patients with bladder carcinoma on immunocompetent cells of the bladder wall. J Urol 144:53–58
Kamat AM, Briggman J, Urbauer DL, Svatek R, Nogueras Gonzalez GM, Anderson R, Grossman HB, Prat F, Dinney CP (2015) Cytokine panel for response to intravesical therapy (CyPRIT): nomogram of changes in urinary cytokine levels predicts patient response to bacillus Calmette–Guerin. Eur Urol. doi:10.1016/jeururo.2015.06.023
Bisiaux A, Thiounn N, Timsit MO, Eladaoui A, Chang HH, Mapes J, Mogenet A, Bresson JL, Prie D, Bechet S, Baron C, Sadorge C, Thomas S, Albert EB, Albert PS, Albert ML (2009) Molecular analyte profiling of the early events and tissue conditioning following intravesical bacillus Calmette–Guerin therapy in patients with superficial bladder cancer. J Urol 181:1571–1580
Biot C, Rentsch CA, Gsponer JR, Birkhauser FD, Jusforgues-Saklani H, Lemaitre F, Auriau C, Bachmann A, Bousso P, Demangel C, Peduto L, Thalmann GN, Albert ML (2012) Preexisting BCG-specific T cells improve intravesical immunotherapy for bladder cancer. Sci Transl Med 4:137ra72
Sonoda T, Sugimura K, Ikemoto S, Kawashima H, Nakatani T (2007) Significance of target cell infection and natural killer cells in the anti-tumor effects of bacillus Calmette–Guerin in murine bladder cancer. Oncol Rep 17:1469–1474
Luo Y, Yamada H, Evanoff DP, Chen X (2006) Role of Th1-stimulating cytokines in bacillus Calmette–Guerin (BCG)-induced macrophage cytotoxicity against mouse bladder cancer MBT-2 cells. Clin Exp Immunol 146:181–188
Suttmann H, Riemensberger J, Bentien G, Schmaltz D, Stockle M, Jocham D, Bohle A, Brandau S (2006) Neutrophil granulocytes are required for effective bacillus Calmette–Guerin immunotherapy of bladder cancer and orchestrate local immune responses. Cancer Res 66:8250–8257
Inman BA, Sebo TJ, Frigola X, Dong H, Bergstralh EJ, Frank I, Fradet Y, Lacombe L, Kwon ED (2007) PD-L1 (B7-H1) expression by urothelial carcinoma of the bladder and BCG-induced granulomata: associations with localized stage progression. Cancer 109:1499–1505
Hurwitz MA, Yao X, Hafez N, Schalper K, Rimm D, Petrylak D (2015) The effect of BCG intravesical therapy and recurrence on PDL1 expression in non-invasive bladder cancers. J Clin Oncol 33:e15504
Carthon BC, Wolchok JD, Yuan J, Kamat A, Ng Tang DS, Sun J, Ku G, Troncoso P, Logothetis CJ, Allison JP, Sharma P (2010) Preoperative CTLA-4 blockade: tolerability and immune monitoring in the setting of a presurgical clinical trial. Clin Cancer Res 16:2861–2871
Kim JW, Bellmunt J, Powles T, Loriot Y, Vogelzang NJ, Zambrano CC, Burris HA, Teng SM, Shen X, Bruey J (2015) Clinical activity, safety, and biomarkers of MPDL3280A in metastatic urothelial bladder cancer: additional analysis from phase IA study. J Clin Oncol (Meeting Abstracts) 33(7)
Petrylak DP, Powles T, Bellmunt J, Braiteh FS, Loriot Y, Cruz Zambrano C, Burris HA, Kim JW, Teng SM, Bruey J (2015) A phase Ia study of MPDL3280A (anti-PDL1): updated response and survival data in urothelial bladder cancer (UBC). J Clin Oncol (Meeting Abstracts) 33
O’Donnell PH, Plimack ER, Bellmunt J, Berger R, Montgomery RB, Heath K, Dolled-Filhart M, Pathiraja K, Gause CK, Cheng JD (2015) Pembrolizumab (Pembro; MK-3475) for advanced urothelial cancer: results of a phase IB study. J Clin Oncol (Meeting Abstracts) 33(7)
Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ (2011) Anti-TIM3 antibody promotes T cell IFN-gamma-mediated antitumor immunity and suppresses established tumors. Cancer Res 71:3540–3551
Jing W, Gershan JA, Weber J, Tlomak D, McOlash L, Sabatos-Peyton C, Johnson BD (2015) Combined immune checkpoint protein blockade and low dose whole body irradiation as immunotherapy for myeloma. J Immunother Cancer 3:2-014-0043-z. eCollection 2015
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27:450–461
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, Duffy SM, Goldberg RM, de la Chapelle A, Koshiji M, Bhaijee F, Huebner T, Hruban RH, Wood LD, Cuka N, Pardoll DM, Papadopoulos N, Kinzler KW, Zhou S, Cornish TC, Taube JM, Anders RA, Eshleman JR, Vogelstein B, Diaz LA Jr (2015) PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med 372:2509–2520
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515:568–571
Herbst RS, Soria J, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN (2014) Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 515:563–567
Liu J, Baras AS, Gandhi NM, Guner G, Munari E, Faraj S, Taube JM, Schoenberg M, Hahn NM, Drake CG (2015) PDL1 status in muscle-invasive urothelial carcinoma in the context of neoadjuvant cisplatin-based chemotherapy. J Clin Oncol (Meeting Abstracts) 33(7)
Wei S, Egenti MU, Teitz-Tennenbaum S, Zou W, Chang AE (2013) Effects of tumor irradiation on host T-regulatory cells and systemic immunity in the context of adoptive T-cell therapy in mice. J Immunother 36:124–132
Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, Deweese TL, Drake CG (2015) Stereotactic radiation therapy augments antigen-specific pd-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res 3:345–355
Vanpouille-Box C, Pilones KA, Wennerberg E, Formenti SC, Demaria S (2015) In situ vaccination by radiotherapy to improve responses to anti-CTLA-4 treatment. Vaccine. doi:10.1016/j.vaccine.2015.05.105
Crittenden M, Kohrt H, Levy R, Jones J, Camphausen K, Dicker A, Demaria S, Formenti S (2015) Current clinical trials testing combinations of immunotherapy and radiation. Semin Radiat Oncol 25:54–64
Ngiow SF, McArthur GA, Smyth MJ (2015) Radiotherapy complements immune checkpoint blockade. Cancer Cell 27:437–438
Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM (2015) Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 520:373–377. doi:10.1038/nature14292
Funding
N.A.S. and M.K.—The Urology Care Foundation, T.J.B.—The Urology Care Foundation, National Institutes of Health
Author contributions
Bivalacqua, Kates, and Sopko contributed to protocol/project development; Bivalacqua, Kates, Sopko, Matsui, Hahn, and Drake contributed to writing and editing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author’s report no conflicts of interest related to this project.
Ethical standards
All ethical standards and obligations were met in carrying out this project.
Rights and permissions
About this article

Cite this article
Kates, M., Sopko, N.A., Matsui, H. et al. Immune checkpoint inhibitors: a new frontier in bladder cancer. World J Urol 34, 49–55 (2016). https://doi.org/10.1007/s00345-015-1709-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00345-015-1709-y