
Abstract
The GATA transcription factors are important transcriptional regulators of plant growth and responses to environmental stimuli. Here, a total of 25 CaGATA genes was identified in chickpea (Cicer arietinum), and their basic characteristics, including gene structure, duplication patterns, conserved domains and various physical and chemical parameters were subsequently determined. Of our interest, the enrichment of the hormone- and stress-responsive cis-regulatory elements in the promoters of CaGATA genes has been analyzed to predict the CaGATA members with potential hormone-mediated functions in stress tolerance. Furthermore, the tissue-specific expression patterns of the CaGATA genes were assessed using the available transcriptome data. More importantly, transcript levels of the identified CaGATA genes were quantified in roots and leaves of chickpea seedlings exposed to ABA (abscisic acid) or dehydration treatment using real-time quantitative PCR. Expression levels of a total of 12 CaGATA genes were significantly altered in roots and/or leaves by both ABA and dehydration treatments, suggesting that these genes might play roles in regulation of chickpea response to water stress in an ABA-dependent manner. Out of these genes, only CaGATA04 was induced in both roots and leaves by ABA and dehydration treatments. Furthermore, CaGATA05 and 21 were the most highly induced in roots (8.55-fold) and leaves (4.90-fold), respectively, by dehydration. Findings of this study have provided important insights into the CaGATA family of chickpea, as well as useful information for selection of CaGATA genes of interest for in-depth functional characterizations that might lead to development of chickpea cultivars with improved performance under water-deficit conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Chickpea (Cicer arietinum) is one of the most important legume crops, which is widely grown in Australia, India, Pakistan, Mexico, and in some Mediterranean and Middle Eastern countries (de Camargo et al. 2019; Muehlbauer and Sarker 2017). Chickpea production has been regarded to be important for sustaining food security owing to the facts that chickpea grains are a primary source of human dietary proteins (El-Beltagi et al. 2017; Gupta et al. 2017), and its capacity for symbiotic nitrogen fixation (Esfahani et al. 2014; Nasr Esfahani et al. 2017). However, chickpea growth, yield, and grain quality are adversely affected by various types of environmental stress, particularly drought (Khodadadi 2013; Kumar et al. 2018).
To cope with water-deficit conditions, plants activate numerous defense mechanisms, starting from stress perception to signal transduction with the involvement of various hormones, and regulatory and functional genes, resulting in activation of a wide range of physiological and biochemical responses to alleviate stress-induced damage (Bagherikia et al. 2019; Kuromori et al. 2018; Li et al. 2019b; Mostofa et al. 2018). In the regulatory networks, various TFs (transcription factors) act as molecular switches to regulate the expression of downstream genes that have drought response-related cis-acting motifs contained in their promoter regions, thereby activating various biological processes in plant acclimation to drought (Hoang et al. 2017; Wang et al. 2018). Increasing evidence has suggested that stress-inducible genes encoding TFs may possess great potential in development of transgenic crops with stable yield under environmental stresses (Chen et al. 2015; Guttikonda et al. 2014; Jin et al. 2016). Among various TF families, the GATA family has been reported to consist of the transcriptional regulatory proteins, which contain a highly conserved type-IV zinc finger as a DNA-binding domain that recognizes promoter elements with a ‘WGATAR’ (W, T/A; R, G/A) core sequence (Behringer and Schwechheimer 2015; Chen et al. 2017; Reyes et al. 2004). Published reports have demonstrated that the GATA TFs not only participate in plant response to low nitrogen stress (Zhang et al. 2015), but also play regulatory roles in plant growth and development (Behringer and Schwechheimer 2015; Jin et al. 2019), such as flowering (Richter et al. 2013), as well as in hormone signaling in plants (Luo et al. 2010). However, the functions of the GATA TFs in chickpea responses to drought and the phytohormone ABA (abscisic acid) have not been well-understood.
In the present study, we carried out a genome-wide analysis to discover all possible CaGATAs (chickpea GATA TF-encoding genes), determined several key features of the GATA TFs, including their gene structure, chromosomal distribution, conserved motifs and phylogenetic relationships, and identified stress- and/or phytohormone-responsive cis-motifs presented in their promoter sequence. The expression patterns of CaGATA genes in different organs during development were also analyzed taking the advantage of available transcriptome atlas. Of our interest, the expression profiles of the identified CaGATA genes in roots and leaves of chickpea seedlings in responses to dehydration and ABA treatments were also investigated using RT-qPCR (real-time quantitative PCR). The obtained data will allow us to identify CaGATA genes that are involved in regulating chickpea response to drought either in ABA-dependent or ABA-independent pathway.
Materials and Methods
Identification and Annotation of CaGATA Genes in Chickpea
All putative CaGATA genes annotated in chickpea were collected from the iTAK database (Plant Transcription factor & Protein Kinase Identifier and Classifier, https://itak.feilab.net/cgi-bin/itak/index.cgi) (Zheng et al. 2016) and the PlantTFDB (Plant Transcription Factor Database, https://planttfdb.cbi.pku.edu.cn) (Jin et al. 2017), following previously published method (Ha et al. 2014). General gene annotation features, consisting of locus identifier, gene identifier, genomic DNA sequence, CDS (coding DNA sequence) and chromosomal location, were retrieved from the chickpea genome assembly (GCF_000331145.1) available at the NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov/assembly/GCF_000331145.1) (Varshney et al. 2013) using BLAST (Basic Local Alignment Search Tool) searches. To confirm the existence of the GATA domain (PF00320) in each CaGATA TF, the full-length protein sequence was queried using the Pfam v. 32.0 (https://pfam.xfam.org) (El-Gebali et al. 2019).
Sequence Analysis for the Gene/Protein Features of the CaGATAs
Gene organization of the putative CaGATAs was analyzed using the GSDS (Gene Structure Display Server) v. 2.0 web-based tool (https://gsds.cbi.pku.edu.cn) (Hu et al. 2015). Chromosomal location of each CaGATA gene was determined using the current genomic sequence of chickpea available at the NCBI RefSeq (Varshney et al. 2013). Protein features of the CaGATA TFs, including theoretical pI (isoelectric point), mW (molecular weight, kDa), instability index and GRAVY (grand average of hydropathy) values, were obtained with the aid of the ExPASy Protparam tool (https://web.expasy.org/protparam) (Artimo et al. 2012). Conserved motif residues in the CaGATA sequences were analyzed using the MEME (Multiple EM for Motif Elicitation) tool (https://meme-suite.org/tools/meme) (Bailey et al. 2015). The parameters were preset as follows: the maximum number of motifs was 15, while the maximum and minimum widths of the motif were 80 and six, respectively.
Phylogenetic Tree Construction and Gene Duplication Analysis
Phylogenetic analysis of the identified CaGATA proteins was performed using the MEGA (Molecular Evolutionary Genetics Analysis, v. 7.0) software (Kumar et al. 2016). Multiple alignment of full-length amino acid sequences of all CaGATA TFs was conducted using ClustalX software (v. 2.1) (Thompson et al. 2002) to attain the conserved GATA domain. The neighbor-joining method was used to build the unrooted phylogenetic tree with the following parameters being applied: 0.2 gap extension penalty, 10.0 gap open penalty, and 1000 replicates for bootstrap analysis. Likewise, the full-length amino acid sequences of all identified AtGATAs and OsGATAs from Arabidopsis thaliana and rice (Oryza sativa), respectively, were obtained from a previous study (Reyes et al. 2004) and used along with the identified CaGATAs to construct a tree for studying their phylogenetic relationship.
The ClustalX was also used for the alignment of the CDSs of the CaGATA genes to assess gene duplication events. Aligned sequence file was exported to the BioEdit (v. 7.2.5) software (Hall 1999) for determination of the identity matrix among the CDSs. The criterion of sharing ≥ 50.0% identity at the nucleotide level was used for defining duplicated genes. Segmental duplications were determined based on the location of duplicated CaGATA genes on different chromosomes, whereas a tandem duplication was declared when duplicated CaGATA genes were located on the same chromosome within a 100-kb distance.
Prediction of the Stress-Related and Phytohormone-Related Cis-acting Motifs
The 1000-bp promoter sequences of all identified CaGATA genes were retrieved from the chickpea genomic sequence (Varshney et al. 2013). A search of the extracted promoter sequences for the well-known light-, stress- and phytohormone-related cis-motifs was conducted using the PlantCARE (Plant Cis-Acting Regulatory Element, https://bioinformatics.psb.ugent.be/webtools/plantcare/html) (Lescot et al. 2002). The distribution of three categories (light-, stress- and phytohormone-responsive) of cis-acting motifs was plotted using TBtools toolkit (Chen et al. 2020).
Expression Analysis of the CaGATA Genes in Different Organs
For organ-specific expression analysis of the identified CaGATA genes in chickpea plants cultivated under normal growth conditions, publically available transcriptome data were obtained from the CTDB (Chickpea Transcriptome Database, https://www.nipgr.res.in/ctdb.html) (Verma et al. 2015). The transcriptome data included (i) the 454 pyrosequencing data that were obtained from roots and shoots of 15-day-old chickpea seedlings grown in culture room, and leaves, flower buds and young pods of mature chickpea plants cultivated under field conditions (Garg et al. 2011), and (ii) the Illumina RNA-sequencing data that were obtained from 5-day-old chickpea seedlings germinated in Petri dishes, shoot apical meristems of 3-week-old chickpea plants cultivated in culture room, and young leaves, flower buds and flowers of chickpea plants cultivated under field conditions at different growth periods (Singh et al. 2013).
Plant Growth and Treatments for RT-qPCR Analysis
Root and leaf samples were collected from chickpea (Hashem ‘kabuli’ cultivar) seedlings, which were grown in pots harboring vermiculite, after they were exposed to dehydration and ABA treatments as described by Ha et al. (2014). Briefly, 9-day-old chickpea plants cultivated on vermiculite were exposed to water (control), 100 μM ABA or dehydration treatment for 2 h and 5 h. The relative water contents in 2-h- and 5-h-dehydrated plant samples were 55% and 33%, respectively (Ha et al. 2014). Leaf and root fractions were then separately collected from the treated chickpea plants for RNA isolation using the RNeasy Plant Mini Kit and QIAcube system (QIAGEN GmbH, Hilden, Germany), DNaseI treatment and cDNA synthesis following the previous procedures (Ha et al. 2014). RT-qPCR was carried out using the Mx3000P qPCR system (Agilent Technologies, Santa Clara, California, USA) (Ha et al. 2014), with IF4a (Initiation factor 4a) (Garg et al. 2010) as a reference gene. Statistically significant differences in changes in gene expression (|fold-change|≥ 1.5) due to treatments were accessed by a Student’s t-test (*P < 0.05, **P < 0.01 and ***P < 0.001). Specific RT-qPCR primers for each CaGATA gene were designed with the aid of Primer 3 (Rozen and Skaletsky 2000) (Online Resource 1).
Results and Discussion
Identification, Annotation and Chromosomal Distribution of the CaGATAs
All potential genes encoding CaGATA TFs (transcription factors) in the ‘kabuli’ chickpea were predicted by the iTAK (Plant Transcription factors & Protein Kinase Identifier and Classifier) (Zheng et al. 2016) and the PlantTFDB (Plant Transcription Factor Database) (Jin et al. 2017) databases. These predicted CaGATA genes were collected from the two databases for sequence matching, which resulted in a list of 25 unique CaGATA members (Table 1). Subsequently, a BLAST (Basic Local Alignment Search Tool) search was conducted by matching these entries against the chickpea genome assembly available at the NCBI (National Center for Biotechnology Information) (Varshney et al. 2013) to obtain the general annotation for each of the identified CaGATA TFs. Chickpea has a lower number of GATA TFs in comparison with A. thaliana, rice and soybean (Glycine max) that have 29, 28 and 64 GATA members, respectively (Reyes et al. 2004; Zhang et al. 2015). Detailed information on the CaGATA genes and CaGATA proteins is given in Table 1, and the nucleotide and aa (amino acid) sequences of each CaGATA are given in Online Resource 2.
The 25 identified CaGATA genes were distributed on eight chromosomes on the basis of the chickpea genome assembly (Varshney et al. 2013), which displayed an unbalanced distribution (Fig. 1). More specifically, six CaGATA genes were mapped on chromosome 7, three CaGATA genes were identified on chromosome 8 and two CaGATA genes were noted on chromosome 3 (Fig. 1). Each of the chromosomes 4, 5 and 6 has four CaGATA genes, while each of chromosomes 1 and 2 possesses one CaGATA gene (Fig. 1). In A. thaliana, three GATA genes were found on each of the chromosomes 1 and 2, while 10, seven and six GATA genes were mapped on chromosomes 3, 4 and 5, respectively (Reyes et al. 2004). Similarly, 28 OsGATA genes were unevenly localized on the rice genome (Reyes et al. 2004). Chromosome 3 has the largest number of OsGATAs (six genes), followed by chromosomes 1, 2 and 5 with four genes per each, whereas each of chromosomes 6, 10, 11 and 12 has two OsGATA genes, and chromosomes 4 and 1 contain only one OsGATA gene (Reyes et al. 2004). Recently, 64 GmGATA genes were unequally mapped on 19 of 20 chromosomes of the soybean genome, excluding chromosome 18. Six GmGATAs were located on chromosome 8, five GmGATAs on each of chromosomes 2, 4, 11, 16 and 17, four GmGATAs on each of chromosomes 6, 7 and 14, three GmGATAs on each of chromosomes 1, 5 and 12, two GmGATAs on each of chromosomes 3, 10, 13, 15 and 19, and one GATA gene on each of chromosomes 9 and 20 (Zhang et al. 2015). These results together suggested that the GATA gene families have multiple members, and the chromosomal distributions of the GATA genes are uneven in chickpea and other plant species.

Chromosomal location of 25 CaGATA genes, and segmental duplication events detected among them. Duplication events detected between two CaGATA genes are shown by red lines, while duplication events detected among three CaGATA genes are shown by green lines. The scale on the left side indicates the length of the chromosomes (Color figure online)
For these identified CaGATAs, their CDSs (coding DNA sequences) were ranged between 402-bp and 1626-bp long, and their corresponding protein lengths were recorded between 133 and 541 aa-s (Table 1). Accordingly, the mWs (molecular weights) of these 25 CaGATA proteins were noticed in the range from 14.9 to 60.2 kDa (Table 1). With respect to their protein stability (Guruprasad et al. 1990), the instability indices of most of the CaGATA proteins were greater than 40, except for that of CaGATA07, 14 and 15, suggesting that only these three proteins are stable by this criterion (Table 1). Theoretical pI (isoelectric point) values were recorded between 4.27 and 10.27, with an average value of 7.40 (Table 1). The GRAVY (grand average of hydropathy) values of all CaGATAs were minus (Table 1), revealing that these CaGATA TFs are hydrophilic. Similar to the protein features of CaGATAs, the length and pI values of GmGATAs were noted to vary greatly, ranging from 80 (9.1 kDa) to 551 aa-s (60.8 kDa) in the protein length, and from 4.63 to 9.66 in the pI values (Zhang et al. 2015). More recently, an investigation of the GATA TFs in grape (Vitis vinifera) also reported a great variation in their protein length, varying from 109 to 386 aa-s (Zhang et al. 2018). These findings collectively suggest that the GATA TFs in various plant species have high variation in size, which might contribute to their functional diversity in plants (Kaur et al. 2018).
Duplication Events of the Members of the CaGATA Family
Gene duplication events, including segmental and tandem duplications, play critical roles in the evolution of a gene family (Jing et al. 2019; Panchy et al. 2016). Thus, we next searched for tandem and segmental duplication events among the annotated CaGATA members using the preset criterion ≥ 50.0% identity. Out of the 25 identified CaGATA genes, eight duplication events comprised 18 CaGATA genes were identified that shared ≥ 50.0% of nucleotide identity, of which six and two duplication events were originated from 12 and six CaGATA genes, respectively (Fig. 1; Online Resource 3). The highest identity of 77.8% was found between CaGATA17 and 23, whereas the lowest identity of 50.0% was noted between CaGATA02 and 15 (two members of the duplication event of three CaGATA genes CaGATA02, 11 and 15) (Fig. 1; Online Resource 3). Due to the different chromosomal locations of all duplicated CaGATA genes involved, all eight duplication events (CaGATA08/10 and 08/20, CaGATA09/16, CaGATA19/25, CaGATA04/22, CaGATA18/24, CaGATA17/23, CaGATA02/11 and 02/15, and CaGATA12/13) were identified as segmental duplications (Fig. 1; Online Resource 3). This finding revealed that these segmental duplications might play the main role in expanding the CaGATA family in chickpea, as also observed with the GATA families in soybean (Zhang et al. 2015) and grape (Zhang et al. 2018). Interestingly, both segmental and whole-genome duplication events were reported to co-occur among the members of the GATA family in apple (Malus domestica) (Chen et al. 2017).
Motif Composition and Gene Structure of the CaGATA Gene Family in Chickpea
Next, to gain an insight into the phylogenetic relationship of the CaGATA TFs, an unrooted phylogenetic tree was built through the alignment of 25 GATA full-length protein sequences (Fig. 2a). According to the created neighbor-joining phylogenetic tree, the CaGATAs could be divided into three groups, namely A, B and C. This classification of the GATA members seems to be conserved in various plant species, at least in chickpea, Arabidopsis and rice (Online Resource 4). Group A contains 17 CaGATA members (68.0%), group B consists of five CaGATA members (20.0%), and group C possesses the lowest number of CaGATAs with three members (12.0%) (Fig. 2a). Results of multiple alignment analysis also revealed that all CaGATA TFs contain the conserved ‘CX2CX18-20CX2C’ type-IV zinc finger domain (Fig. 2b). This domain has the conserved motif ‘LCNACG’ spanning the second cysteine pair, and the ‘TPQWR’, ‘TPLWR’, ‘TPAMR’, ‘TPMMR’ or ‘APHWR’ motif within the X18–20 loop (Fig. 2b). The CaGATA members in group A contain the ‘TPQWR’ or ‘TPLWR’ motif, while those of group C consist of ‘TPLWR’ or ‘APHWR’ motif within the 18-residue loop (Fig. 2a, b). The five CaGATA members of group B contain ‘TPAMR’ or ‘TPMMR’ motif within the 20-residue loop (Fig. 2a, b). Similar observations were also noted for the members of the groups A, B and C in other species like Arabidopsis and rice (Reyes et al. 2004), soybean (Zhang et al. 2015), apple (Chen et al. 2017) and grape (Zhang et al. 2018).

Phylogenetic tree of 25 CaGATA proteins, schematic distribution of the conserved amino acid motifs among the CaGATA proteins, and schematic presentation of exon and intron distributions in the CaGATA genes. a The tree was created using the full-length sequences of 25 identified CaGATA TFs. b Alignment of the GATA domain (motif 1) obtained from 25 CaGATA TFs. c Motif compositions of the CaGATA TFs in chickpea. The motifs, numbered 1–15, are shown in different colored boxes. The sequence information for each motif is listed in Online Resource 5. The protein length in amino acids can be estimated using the scale below the image. d Exon–intron structures of CaGATA genes. Yellow boxes represent exons, blue boxes indicate upstream sequences and black lines indicate introns. The gene size can be estimated using the scale below the image (Color figure online)
For further analysis of the motif compositions within the CaGATA TFs, we used the MEME (Multiple EM for Motif Elicitation) software (Bailey et al. 2015) to explore the diversity of the motifs in each CaGATA protein (Fig. 2c). A total of 15 different motifs with various aa lengths and sequences were detected in the chickpea CaGATA TFs, which were named motifs 1–15 (Fig. 2c; Online Resource 5). All CaGATA TFs contained the typical type-IV zinc finger (motif 1) (Fig. 2b, c; Online Resource 5). It is worth mentioning that the group-B CaGATA TFs possess two additional well-known motifs (out of the total four motifs), representing the TIFY (motif 3) and the CCT domain (motif 5) (Fig. 2a, c; Online Resource 5). TIFY domain-containing proteins (with a core motif ‘TLS[F/Y]XG’) have been established to be implicated in jasmonic acid-related stress responses and developmental processes (Bai et al. 2011; Vanholme et al. 2007). The CCT domain was first discovered in CONSTANS (flowering time controller proteins), CONSTANS-like (Griffiths et al. 2003) and TOC1 (timing of cab expression 1) proteins of Arabidopsis (Strayer et al. 2000), which were shown to play important roles in photoperiodic signaling, and mediate protein–protein interactions in Arabidopsis plants (Robson et al. 2001; Strayer et al. 2000). Some group-B GATA TFs of Arabidopsis and rice plants (Reyes et al. 2004) (Online Resource 4), as well as several group-C GATA TFs in apple (Chen et al. 2017), also possess the TIFY and CCT motifs.
The diversity of gene structure may be a mechanism to promote the evolution of multigenic families (Roy and Gilbert 2006). The structures of all 25 CaGATA genes are shown in Fig. 2d, in which the members within each group displayed similar exon/intron structures (Fig. 2a, d). Group A has the largest number of CaGATA genes with two or three exons (Fig. 2a, d). Members in group B possess seven, eight, 10 or 11 exons, while those in group C contain seven exons, except CaGATA14 that has three exons (Fig. 2a, d). These gene structures of the identified CaGATAs were found to be similar to those of Arabidopsis, rice (Reyes et al. 2004) and soybean (Zhang et al. 2015). These results collectively suggested that the members of the same group in the phylogenetic tree may share similar motif composition and gene structure (Fig. 2a–d). The differences in motif composition and gene structure may contribute, at least partly, to the diverse functions of the GATA TFs (Kaur et al. 2018; Li et al. 2019a).
Enrichment Analysis of Putative Cis-acting Motifs in the Promoter Sequences of CaGATA Genes
The distribution of cis-motifs may reflect the potential transcriptional regulation and functions of genes (Walley et al. 2007). To obtain an insight into the signaling functions of all 25 identified CaGATA genes, the 1000-bp upstream sequence of each CaGATA gene was retrieved, as promoter region of each gene, from the available genome assembly of chickpea (Varshney et al. 2013). The obtained promoter sequences were then analyzed for the occurrences of cis-acting motifs with the aid of the PlantCARE (Plant Cis-Acting Regulatory Element) database (Lescot et al. 2002). Besides the core motifs, such as the TATA-box and CAAT-box, which form a common feature in most of the eukaryotic promoters (Zhu et al. 1995), a total of 33 types of cis-motifs were discovered (Fig. 3). Specifically, our results revealed 19 light-responsive motifs in the promoter sequences of all CaGATAs, especially the Box 4 element (Jeong and Shih 2003) that showed 64 out of 161 occurrences distributed within the 1000-bp promoter sequences of 23 out of 25 CaGATA genes (Fig. 3). Following the Box 4 element, 28 and 18 occurrences of the light-responsive G box and GATA motif (Jeong and Shih 2003), respectively, were found in the promoters of 11 and 13 (out of 25) CaGATA genes (Fig. 3). Results of this in silico analysis also revealed that most of the CaGATA genes contain light-responsive cis-acting regulatory elements, except CaGATA08, with CaGATA20 and 21 sharing the highest number of total occurrence of 12 (Fig. 3).

Analysis for the occurrences of well-known light-, stress- and hormone-responsive cis-motifs in the promoter regions of CaGATA genes. Heatmap represents the occurrences of cis-motifs in the promoter sequences. Cis-motifs and their occurrences in the 1000-bp promoter sequences were predicted using the PlantCARE server. The 1000-bp regions upstream of the start codon of each gene were extracted from the corresponding genomic sequence. The color gradient indicates the occurrences of each motif in the promoter regions (Color figure online)
Of our interest, some stress- and phytohormone-responsive cis-motifs were detected in the promoter sequences of the identified CaGATAs (Fig. 3). Stress-responsive cis-motifs like the EE (evening element), MBS (MYB-binding site), DRE (dehydration-responsive element), MYCR (MYC-binding site), NACR (binding site of NAC TFs), WUN-motif (wound-responsive element), CE3 (coupling element 3), LTRE (low temperature-responsive element) and TC-rich repeats have been recognized as dehydration/drought-inducible cis-motifs (Abe et al. 2003; Maruyama et al. 2012; Ramya et al. 2010; Tran et al. 2004; Yamaguchi-Shinozaki and Shinozaki 2005), which are involved in mediating the drought-responsive transcriptional regulation of many genes in plants (Hoang et al. 2017; Hussain et al. 2017). In this study, MBS, MYCR, LTRE, WUN-motif and/or TC-rich repeats cis-motifs were identified in the promoter regions of 12 out of 25 CaGATA genes. In addition, several phytohormone-responsive elements, including ERE (ethylene-responsive element), ABRE (ABA-responsive element), TATC-box, GARE-motif and P-box (gibberellin-responsive element), TGA-element (auxin-responsive element), TCA-element (salicylic acid-responsive element) and/or methyl jasmonate-responsive motifs (e.g., motifs ‘TGACG’ and ‘CGTCA’), were also found in the 1000-bp promoter sequences of most of the CaGATA genes (23 out of 25), except CaGATA06 and 25 (Fig. 3). It is worth mentioning that the promoters of some CaGATA genes (12 out of 25) possess at least one stress-responsive motif (Fig. 3). For instance, CaGATA01, 03 and 04 contain at least one MYCR and one ABRE, implying that these CaGATAs might be involved in regulating plant response to drought/dehydration in an ABA-dependent manner (Nakashima and Yamaguchi-Shinozaki 2013; Yamaguchi-Shinozaki and Shinozaki 2005). In short, our cis-motif enrichment analysis indicated that the identified CaGATAs might control a wide range of biological processes in chickpea, including its responses to different types of environmental stress.
Expression Patterns of CaGATA Genes in Several Organs During Chickpea Growth and Development
To understand the expression patterns of the identified CaGATA genes, and their functional divergence on the basis of their expression profiles, we next conducted a re-analysis of the transcriptome data obtained from different organs, including shoots, roots, young leaves, mature leaves, flower buds, young pods of chickpea, which are available at the CTDB (Chickpea Transcriptome Database) (Verma et al. 2015). Among the identified CaGATAs, six genes (CaGATA04, 08, 10, 14, 18 and 24) had no information on their expression from the reported transcriptome data (Garg et al. 2011; Singh et al. 2013), while CaGATA19 showed very low transcript levels (Fig. 4). According to the 454 pyrosequencing data (Garg et al. 2011), 18 CaGATA genes displayed differential transcription patterns in the investigated organs (Fig. 4). For example, when comparing the expression patterns of CaGATAs in the examined organs, CaGATA09 showed the highest transcript level in roots, while CaGATA21 and 25 were the most strongly expressed genes in shoots and flower buds, respectively, than in other organs (Fig. 4a). The transcription profiles of CaGATAs in various organs collected at different developmental stages were also documented with the aid of the RNA-sequencing data produced by Singh et al. (2013) using the Illumina platform. Heatmap presented in Fig. 4b showed that the majority of the CaGATA genes displayed differential transcription patterns in the investigated organs, especially the CaGATAs of group A. For example, CaGATA05 and 21 displayed high transcription patterns in young leaves, while CaGATA09 exhibited a high expression level in shoot apical meristem (Fig. 4b). Taken together, our re-analysis of the published transcriptome sources to assess the expression profiles of the identified CaGATA genes allowed us to make a concluding remark that the encoding CaGATA TFs have diverse functions in controlling the development of various organs in chickpea plants during their life circle.

Heatmap of CaGATA gene expression levels in different organs at different growth periods of chickpea. Data used in the analysis were collected from the Chickpea Transcriptome Database. a Expression patterns of identified CaGATA genes according to the 454 pyrosequencing data. b Expression patterns of identified CaGATA genes according to the Illumina RNA-sequencing data. Germinating seedlings, GS; young leaves, YL; shoot apical meristem, SAM; flower bud stages 1–4, FB1-FB4; flower stages 1–4, FL1-FL4. Color scales indicate transcript levels according to reads per million values (a), and log2 basemean values (b) (Color figure online)
Transcription Profiles of the CaGATA Genes in Roots and Leaves of the Chickpea Seedlings Exposed to Dehydration and Exogenous ABA Treatments
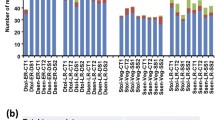
To gain a better understanding regarding the potential functions of the CaGATA genes in chickpea response to drought, we next used RT-qPCR (real-time quantitative PCR) to examine their expression profiles in roots and leaves of chickpea seedlings exposed to dehydration, as well as exogenous ABA treatment since ABA has been well established as a key hormone regulating plant drought responses (Kuromori et al. 2018; Osakabe et al. 2014a, b). As shown in Figs. 5 and 6, a number of the identified CaGATA genes exhibited differential transcript levels in roots and/or leaves in response to dehydration. Ten genes (CaGATA04, 05, 06, 07, 10, 18, 19, 20, 21 and 24) were upregulated by a minimum of 1.5-fold (P-value < 0.05) in roots (six genes, CaGATA04, 05, 06, 10, 21 and 24) and/or leaves (seven genes, CaGATA04, 07, 18, 19, 20, 21 and 24) of chickpea plants treated with dehydration (Figs. 5 and 6; Online Resource 6). For instance, CaGATA05 and 21 were the most highly induced in roots (~ 8.55-fold) (Fig. 5; Online Resource 6), and leaves (~ 4.90-fold), respectively, by dehydration (Fig. 6; Online Resource 6). Furthermore, CaGATA21 was upregulated by the highest levels in both dehydrated roots (~ 2.83-fold) and leaves (~ 4.90-fold) (Figs. 5 and 6; Online Resource 6). In contrast, CaGATA16 was the most highly repressed gene by dehydration in both roots (~ 4.01-fold) and leaves (~ 5.75-fold) (Figs. 5 and 6). Intriguingly, CaGATA07, 18 and 20 were noted to be induced after 2 h, but they were then repressed after 5 h of dehydration in leaves (Figs. 5 and 6; Online Resource 6).

Transcriptional responses of CaGATA genes in chickpea roots under ABA and dehydration treatments. Mean relative expression levels of each CaGATA gene were normalized to a value of 1 in the respective water-treated control. Error bars indicate the SE values of three biological replicates (n = 3). Asterisks show significant differences in expression changes as assessed by a Student's t-test (|fold-change|≥ 1.5; *P < 0.05, **P < 0.01, ***P < 0.001)

Transcriptional responses of CaGATA genes in chickpea leaves under ABA and dehydration treatments. Mean relative expression levels of each CaGATA gene were normalized to a value of 1 in the respective water-treated control. Error bars indicate the SE values of three biological replicates (n = 3). Asterisks show significant differences in expression changes as assessed by a Student's t-test (|fold-change|≥ 1.5; *P < 0.05, **P < 0.01, ***P < 0.001)
With respect to the ABA treatment, nine genes (CaGATA01, 03, 04, 06, 12, 14, 19, 21 and 24) were upregulated in roots (five genes, CaGATA01, 04, 06, 12 and 19) and/or leaves (seven genes, CaGATA03, 04, 06, 14, 19, 21 and 24), whereas six genes (CaGATA09, 10, 16, 17, 20 and 24) were downregulated by a minimum of 1.5-fold (P-value < 0.05) in roots (six genes, CaGATA09, 10, 16, 17, 20 and 24) and/or leaves (three genes, CaGATA09, 16 and 20) (Figs. 5 and 6; Online Resource 6). Additionally, several stress-responsive and/or hormone-responsive cis-motifs were detected in the promoter sequences of these dehydration-responsive and/or ABA-responsive CaGATAs (Fig. 3), supporting our RT-qPCR results (Figs. 5 and 6). Interestingly, the majority of the ABA-responsive CaGATA genes (12 out of 14), including CaGATA01, 04, 06, 09, 10, 12, 16, 17, 19, 20, 21 and 24, were also found to be dehydration-responsive in roots and/or leaves (Figs. 5 and 6; Online Resource 6). These data suggested that these CaGATA genes might be involved in ABA-dependent regulation of chickpea response to water scarcity. It is also worth mentioning that CaGATA04 was induced in both chickpea roots and leaves by ABA and dehydration treatments.
Conclusions
In this study, 25 CaGATA genes were identified, and their classification and evolutionary relationships were subsequently assessed via a series of analyses, including phylogenetic, gene structure and conserved protein motif analyses. The identified CaGATA genes displayed different transcription patterns in different organs of chickpea plants across developmental stages, as well as under the conditions of dehydration and ABA treatments. Findings also demonstrated that the expression of 12 CaGATA genes is modulated by dehydration through ABA-dependent pathway in roots and/or leaves of chickpea plants. CaGATA05 and CaGATA21 were the most highly induced in roots and leaves, respectively, by dehydration; and thus, they could be used for improving drought tolerance with specific attention to root and leaf organs. In addition, CaGATA04 was induced by both dehydration and ABA treatments in both roots and leaves, which would be also an interesting candidate for in-depth functional analyses in the future. Findings of this study will collectively contribute to the understanding of potential roles of CaGATA genes in regulating the responses of chickpea plants to water-deficit stress and ABA treatment, and enable us to select promising genes for genetic engineering with the aim of developing chickpea varieties with improved productivity under drought.
References
Abe H, Urao T, Ito T, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. Plant Cell 15(1):63–78. https://doi.org/10.1105/tpc.006130
Artimo P et al (2012) ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res 40:W597–603. https://doi.org/10.1093/nar/gks400
Bagherikia S, Pahlevani M, Yamchi A, Zaynalinezhad K, Mostafaie A (2019) Transcript profiling of genes encoding fructan and sucrose metabolism in wheat under terminal drought stress. J Plant Growth Regul 38:148–163. https://doi.org/10.1007/s00344-018-9822-y
Bai Y, Meng Y, Huang D, Qi Y, Chen M (2011) Origin and evolutionary analysis of the plant-specific TIFY transcription factor family. Genomics 98(2):128–136. https://doi.org/10.1016/j.ygeno.2011.05.002
Bailey TL, Johnson J, Grant CE, Noble WS (2015) The MEME suite. Nucleic Acids Res 43(W1):W39–W49. https://doi.org/10.1093/nar/gkv416
Behringer C, Schwechheimer C (2015) B-GATA transcription factors—insights into their structure, regulation, and role in plant development. Front Plant Sci 6:90. https://doi.org/10.3389/fpls.2015.00090
Chen M, Zhao Y, Zhuo C, Lu S, Guo Z (2015) Overexpression of a NF-YC transcription factor from bermudagrass confers tolerance to drought and salinity in transgenic rice. Plant Biotechnol J 13(4):482–491. https://doi.org/10.1111/pbi.12270
Chen H, Shao H, Li K, Zhang D, Fan S, Li Y, Han M (2017) Genome-wide identification, evolution, and expression analysis of GATA transcription factors in apple (Malusx domestica Borkh.). Gene 627:460–472. https://doi.org/10.1016/j.gene.2017.06.049
Chen C, Chen H, Zhang Y, Thomas HR, Frank MH, He Y, Xia R (2020) TBtools, a toolkit for biologists integrating various biological data handling tools with a user-friendly interface. bioRxiv. https://doi.org/10.1101/289660
de Camargo AC et al (2019) Is chickpea a potential substitute for soybean? Phenolic bioactives and potential health benefits. Int J Mol Sci 20(11):E2644. https://doi.org/10.3390/ijms20112644
El-Beltagi HS, El-Senousi NA, Ali ZA, Omran AA (2017) The impact of using chickpea flour and dried carp fish powder on pizza quality. PLoS ONE 12(9):e0183657. https://doi.org/10.1371/journal.pone.0183657
El-Gebali S et al (2019) The Pfam protein families database in 2019. Nucleic Acids Res 47(Database issue):D427–D432. https://doi.org/10.1093/nar/gky995
Esfahani MN, Sulieman S, Schulze J, Yamaguchi-Shinozaki K, Shinozaki K, Tran LS (2014) Approaches for enhancement of N(2) fixation efficiency of chickpea (Cicer arietinum L.) under limiting nitrogen conditions. Plant Biotechnol J 12:387–397. https://doi.org/10.1111/pbi.12146
Garg R, Sahoo A, Tyagi AK, Jain M (2010) Validation of internal control genes for quantitative gene expression studies in chickpea (Cicer arietinum L.). Biochem Biophys Res Commun 396(2):283–288. https://doi.org/10.1016/j.bbrc.2010.04.079
Garg R et al (2011) Gene discovery and tissue-specific transcriptome analysis in chickpea with massively parallel pyrosequencing and web resource development. Plant Physiol 156(4):1661–1678. https://doi.org/10.1104/pp.111.178616
Griffiths S, Dunford RP, Coupland G, Laurie DA (2003) The evolution of CONSTANS-like gene families in barley, rice, and Arabidopsis. Plant Physiol 131(4):1855–1867. https://doi.org/10.1104/pp.102.016188
Gupta RK, Gupta K, Sharma A, Das M, Ansari IA, Dwivedi PD (2017) Health risks and benefits of chickpea (Cicer arietinum) consumption. J Agric Food Chem 65(1):6–22. https://doi.org/10.1021/acs.jafc.6b02629
Guruprasad K, Reddy BV, Pandit MW (1990) Correlation between stability of a protein and its dipeptide composition: a novel approach for predicting in vivo stability of a protein from its primary sequence. Protein Eng 4(2):155–161. https://doi.org/10.1093/protein/4.2.155
Guttikonda SK et al (2014) Overexpression of AtDREB1D transcription factor improves drought tolerance in soybean. Mol Biol Rep 41(12):7995–8008. https://doi.org/10.1007/s11033-014-3695-3
Ha CV et al (2014) Genome-wide identification and expression analysis of the CaNAC family members in chickpea during development, dehydration and ABA treatments. PLoS ONE 9(12):e114107. https://doi.org/10.1371/journal.pone.0114107
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hoang XLT, Nhi DNH, Thu NBA, Thao NP, Tran LP (2017) Transcription factors and their roles in signal transduction in plants under abiotic stresses. Curr Genomics 18(6):483–497. https://doi.org/10.2174/1389202918666170227150057
Hu B, Jin J, Guo AY, Zhang H, Luo J, Gao G (2015) GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31(8):1296–1297. https://doi.org/10.1093/bioinformatics/btu817
Hussain RM, Ali M, Feng X, Li X (2017) The essence of NAC gene family to the cultivation of drought-resistant soybean (Glycine max L. Merr.) cultivars. BMC Plant Biol 17(1):55. https://doi.org/10.1186/s12870-017-1001-y
Jeong MJ, Shih MC (2003) Interaction of a GATA factor with cis-acting elements involved in light regulation of nuclear genes encoding chloroplast glyceraldehyde-3-phosphate dehydrogenase in Arabidopsis. Biochem Biophys Res Commun 300(2):555–562. https://doi.org/10.1016/s0006-291x(02)02892-9
Jin C, Huang XS, Li KQ, Yin H, Li LT, Yao ZH, Zhang SL (2016) Overexpression of a bHLH1 transcription factor of Pyrus ussuriensis confers enhanced cold tolerance and increases expression of stress-responsive genes. Front Plant Sci 7:441. https://doi.org/10.3389/fpls.2016.00441
Jin J, Tian F, Yang D-C, Meng Y-Q, Kong L, Luo J, Gao G (2017) PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res 45(D1):D1040–D1045. https://doi.org/10.1093/nar/gkw982
Jin J et al (2019) The transcription factor GATA10 regulates fertility conversion of a two-line hybrid tms5 mutant rice via the modulation of UbL40 expression. J Integr Plant Biol. https://doi.org/10.1111/jipb.12871
Jing X-Q et al (2019) Genome-wide identification of malectin/malectin-like domain containing protein family genes in rice and their expression regulation under various hormones, abiotic stresses, and heavy metal treatments. J Plant Growth Regul 39:492–506. https://doi.org/10.1007/s00344-019-09997-8
Kaur G, Guruprasad K, Temple BRS, Shirvanyants DG, Dokholyan NV, Pati PK (2018) Structural complexity and functional diversity of plant NADPH oxidases. Amino Acids 50(1):79–94. https://doi.org/10.1007/s00726-017-2491-5
Khodadadi M (2013) Effect of drought stress on yield and water relative content in chickpea. Int J Agil Agron Plant Prod 4:1168–1172
Kumar S, Stecher G, Tamura K (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol 33(7):1870–1874. https://doi.org/10.1093/molbev/msw054
Kumar M, Yusuf MA, Nigam M, Kumar M (2018) An update on genetic modification of chickpea for increased yield and stress tolerance. Mol Biotechnol 60(8):651–663. https://doi.org/10.1007/s12033-018-0096-1
Kuromori T, Seo M, Shinozaki K (2018) ABA transport and plant water stress responses. Trends Plant Sci 23(6):513–522. https://doi.org/10.1016/j.tplants.2018.04.001
Lescot M et al (2002) PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res 30(1):325–327. https://doi.org/10.1093/nar/30.1.325
Li D, Wu D, Li S, Dai Y, Cao Y (2019a) Evolutionary and functional analysis of the plant-specific NADPH oxidase gene family in Brassica rapa L. R Soc Open Sci 6(2):181727. https://doi.org/10.1098/rsos.181727
Li W, Herrera-Estrella L, Tran L-SP (2019b) Do cytokinins and strigolactones crosstalk during drought adaptation? Trends Plant Sci 24(8):669–672. https://doi.org/10.1016/j.tplants.2019.06.007
Luo XM et al (2010) Integration of light- and brassinosteroid-signaling pathways by a GATA transcription factor in Arabidopsis. Dev Cell 19(6):872–883. https://doi.org/10.1016/j.devcel.2010.10.023
Maruyama K et al (2012) Identification of cis-acting promoter elements in cold- and dehydration-induced transcriptional pathways in Arabidopsis, rice, and soybean. DNA Res 19(1):37–49. https://doi.org/10.1093/dnares/dsr040
Mostofa MG, Li W, Nguyen KH, Fujita M, Tran LP (2018) Strigolactones in plant adaptation to abiotic stresses: an emerging avenue of plant research. Plant Cell Environ 41(10):2227–2243. https://doi.org/10.1111/pce.13364
Muehlbauer FJ, Sarker A (2017) Economic importance of chickpea: production, value, and world trade. In: Varshney RK, Thudi M, Muehlbauer F (eds) The chickpea genome. Springer International Publishing, Cham, pp 5–12. https://doi.org/10.1007/978-3-319-66117-9_2
Nakashima K, Yamaguchi-Shinozaki K (2013) ABA signaling in stress-response and seed development. Plant Cell Rep 32(7):959–970. https://doi.org/10.1007/s00299-013-1418-1
Nasr Esfahani M et al (2017) Comparative transcriptome analysis of nodules of two Mesorhizobium-chickpea associations with differential symbiotic efficiency under phosphate deficiency. Plant J 91(5):911–926. https://doi.org/10.1111/tpj.13616
Osakabe Y, Osakabe K, Shinozaki K, Tran L-S (2014a) Response of plants to water stress. Front Plant Sci 5:86. https://doi.org/10.3389/fpls.2014.00086
Osakabe Y, Yamaguchi-Shinozaki K, Shinozaki K, Tran LS (2014b) ABA control of plant macroelement membrane transport systems in response to water deficit and high salinity. New Phytol 202(1):35–49. https://doi.org/10.1111/nph.12613
Panchy N, Lehti-Shiu M, Shiu S-H (2016) Evolution of gene duplication in plants. Plant Physiol 171(4):2294–2316. https://doi.org/10.1104/pp.16.00523
Ramya M, Raveendran M, Sathiyamoorthy S, Ramalingam J (2010) In silico analysis of drought tolerant genes in rice. Int J Biol Med Res 1(3):36–40
Reyes JC, Muro-Pastor MI, Florencio FJ (2004) The GATA family of transcription factors in Arabidopsis and rice. Plant Physiol 134(4):1718–1732. https://doi.org/10.1104/pp.103.037788
Richter R, Bastakis E, Schwechheimer C (2013) Cross-repressive interactions between SOC1 and the GATAs GNC and GNL/CGA1 in the control of greening, cold tolerance, and flowering time in Arabidopsis. Plant Physiol 162(4):1992–2004. https://doi.org/10.1104/pp.113.219238
Robson F et al (2001) Functional importance of conserved domains in the flowering-time gene CONSTANS demonstrated by analysis of mutant alleles and transgenic plants. Plant J 28(6):619–631. https://doi.org/10.1046/j.1365-313x.2001.01163.x
Roy SW, Gilbert W (2006) The evolution of spliceosomal introns: patterns, puzzles and progress. Nat Rev Genet 7(3):211–221. https://doi.org/10.1038/nrg1807
Rozen S, Skaletsky H (2000) Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol 132:365–386. https://doi.org/10.1385/1-59259-192-2:365
Singh VK, Garg R, Jain M (2013) A global view of transcriptome dynamics during flower development in chickpea by deep sequencing. Plant Biotechnol J 11(6):691–701. https://doi.org/10.1111/pbi.12059
Strayer C et al (2000) Cloning of the Arabidopsis clock gene TOC1, an autoregulatory response regulator homolog. Science 289(5480):768–771. https://doi.org/10.1126/science.289.5480.768
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinform. https://doi.org/10.1002/0471250953.bi0203s00
Tran LS et al (2004) Isolation and functional analysis of Arabidopsis stress-inducible NAC transcription factors that bind to a drought-responsive cis-element in the early responsive to dehydration stress 1 promoter. Plant Cell 16(9):2481–2498. https://doi.org/10.1105/tpc.104.022699
Vanholme B, Grunewald W, Bateman A, Kohchi T, Gheysen G (2007) The tify family previously known as ZIM. Trends Plant Sci 12(6):239–244. https://doi.org/10.1016/j.tplants.2007.04.004
Varshney RK et al (2013) Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31(3):240–246. https://doi.org/10.1038/nbt.2491
Verma M, Kumar V, Patel RK, Garg R, Jain M (2015) CTDB: an integrated chickpea transcriptome database for functional and applied genomics. PLoS ONE 10(8):e0136880. https://doi.org/10.1371/journal.pone.0136880
Walley JW et al (2007) Mechanical stress induces biotic and abiotic stress responses via a novel cis-element. PLoS Genet 3(10):1800–1812. https://doi.org/10.1371/journal.pgen.0030172
Wang C et al (2018) Auxin is involved in lateral root formation induced by drought stress in tobacco seedlings. J Plant Growth Regul 37:539–549. https://doi.org/10.1007/s00344-017-9752-0
Yamaguchi-Shinozaki K, Shinozaki K (2005) Organization of cis-acting regulatory elements in osmotic- and cold-stress-responsive promoters. Trends Plant Sci 10(2):88–94. https://doi.org/10.1016/j.tplants.2004.12.012
Zhang C et al (2015) Genome-wide survey of the soybean GATA transcription factor gene family and expression analysis under low nitrogen stress. PLoS ONE 10(4):e0125174. https://doi.org/10.1371/journal.pone.0125174
Zhang Z, Ren C, Zou L, Wang Y, Li S, Liang Z (2018) Characterization of the GATA gene family in Vitis vinifera: genome-wide analysis, expression profiles, and involvement in light and phytohormone response. Genome 61(10):713–723. https://doi.org/10.1139/gen-2018-0042
Zheng Y et al (2016) iTAK: a program for genome-wide prediction and classification of plant transcription factors, transcriptional regulators, and protein kinases. Mol Plant 9(12):1667–1670. https://doi.org/10.1016/j.molp.2016.09.014
Zhu Q, Dabi T, Lamb C (1995) TATA box and initiator functions in the accurate transcription of a plant minimal promoter in vitro. Plant Cell 7(10):1681–1689. https://doi.org/10.1105/tpc.7.10.1681
Acknowledgements
Cuong Duy Tran would like to acknowledge the PhD fellowship funded by the International Program Associate of RIKEN.
Author information
Authors and Affiliations
Contributions
Conceptualization: L-SPT; Methodology: LN, HDC, L-SPT; Formal analysis and investigation: LN, HDC, CDT, KHN, HXP, DTL, WL, WW, L-SPT; Writing—original draft preparation: LN, HDC, L-SPT; Writing—review and editing: LN, HDC, TDL, L-SPT; Resources: TDL, L-SPT; Supervision: TDL, L-SPT.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Niu, L., Chu, H.D., Tran, C.D. et al. The GATA Gene Family in Chickpea: Structure Analysis and Transcriptional Responses to Abscisic Acid and Dehydration Treatments Revealed Potential Genes Involved in Drought Adaptation. J Plant Growth Regul 39, 1647–1660 (2020). https://doi.org/10.1007/s00344-020-10201-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-020-10201-5