
Abstract
We employed a hybrid femtosecond/picosecond coherent anti-Stokes Raman scattering (fs/ps CARS) instrument to probe rotational temperatures of molecular hydrogen in the multiphase reaction zone of an aluminized ammonium perchlorate (AP) propellant flame. Significant concentrations of hydrogen, present in the plume due to the decomposition of the propellant binder material and subsequent reactions with AP oxidizers, allowed for single-shot thermometry at the laser repetition rate of 1 kHz. A time-asymmetric picosecond probe pulse time-gated the impulsively generated Raman coherence at a delay of 2.66 ps from the pump and Stokes pulses, before any appreciable coherence dephasing occurred in the atmospheric pressure flames and mitigating uncertainties in the Raman transition frequencies and dephasing processes. Measurements in near-adiabatic H2-air flames at equivalence ratios of \(\phi\) = 1–1.8 demonstrated measurement accuracy to near 5% of equilibrium predictions with a precision approaching 3% for high signal-to-noise ratio spectra. Introduction of a time-delayed probe pulse provided Raman-resonant spectra from the plumes of burning propellants with ample signal above broadband background emission, which were fit to libraries of synthetic spectra to infer the gas rotational temperature 0–15 mm from the burning surface. The mean fitted temperature of 2494 K from three propellant burns compares favorably to other measurements of gas and particle temperatures in similar propellant studies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
While solid propellants are an integral component of rocket propulsion, there remains much to learn about the burn characteristics of these propellants, especially at off-design conditions experienced under an accident scenario where the burns may occur at or near atmospheric pressure [1]. Improved modeling efforts over the years have continued to capture more of the underlying physics [2,3,4,5], but there remains a need for experimental validation data. However, propellant burns provide a diagnostic challenge, featuring high temperatures, intense background emission, complex chemistry, and often multi-phase plumes due to the addition of metal particles (typically aluminum) which are added to increase temperature and specific impulse [6]. Early spectroscopic work, summarized by Edwards [5], focused efforts on metal-free propellants where a variety of non-intrusive laser-based diagnostics were of use, including laser induced florescence (LIF), spontaneous Raman scattering, IR absorption, and coherent anti-Stokes Raman scattering (CARS) (see table 1 in Ref. [5]). However, the addition of aluminum particulate initially limited diagnostics to emission imaging above the background from the broad aluminum particle spectrum of burns near atmospheric pressure.
Significant diagnostic development has led to successful measurements in these metal-particle-laden burning environments, with demonstrations of planar LIF [7]; digital in-line holography (DIH) for particle tracking and size [8]; DIH with two-color pyrometry for aluminum particle temperatures [9]; CARS for rotational temperatures and relative O2/N2 concentrations [10]; and laser-induced breakdown spectroscopy (LIBS) for aluminum vapor concentration measurements [11]. Previous work from our lab involving ultrafast CARS probing of nitrogen and oxygen rotational states [10] demonstrated the advantage of introducing a picosecond CARS probe pulse at a time delay from femtosecond pump and Stokes pulses for mitigation of nonresonant background contamination that has plagued traditional nanosecond CARS in environments with large, micron-scale particles [12]. However, rotational CARS detection of N2 and O2 in Ref. [10] was subject to spatial averaging of combustion gases with air entrained into the propellant flame, potentially providing a cold bias to the fitted spectra. In addition, while single-laser-shot measurements were performed, the signal strengths were less than desired as a result of very high temperatures and low concentrations of both N2 and O2 in the propellant gas plume, suggesting the need to examine another species for gas-phase thermometry purposes.
Our lab has focused specifically on small benchtop burns of ammonium perchlorate (AP—NH4ClO4), hydroxyl terminated polybutadiene (HTPB, a molecular complex of butadiene—C4H6, bounded by OH), and aluminum (AL) composites like the one featured in Fig. 1a, with a propellant formulation that is nominally 70% AP, 20% AL, and 10% HTPB by weight. Considering only the AP and the HTPB binder for now, the resulting flame structure after ignition is typically thought of as a combination of three flames [2, 4, 5, 13] described in Eqs. 1–3 and depicted in Fig. 1b. The first flame area is a monopropellant flame as the oxidizer (HClOx, ClOx, etc.) and fuel (NH3) portions of AP thermal decomposition react. The second, or “primary,” flame zone is formed as the decomposed AP oxidizers react with the fuel-rich products of thermal decomposition of the binder (CH3, H2, etc.). This flame occurs on the physical boundaries of the binder and AP at the burning surface. The third, or “final”, diffusion flame zone consists of reactions from the remaining fuel species of the binder and any products from the first two flames. These reactions are described as follows, where Px denotes the products for reaction x, with finer details on the complex chemistry found elsewhere [14, 15]:

a Example image of the propellant stick during burn. For reference, the adjacent lens has a 50-mm diameter. b Cartoon of a propellant burning surface, detailing the three-flame model for AP/HTPB combustion
P1 consists of oxidizing and inert species at or near temperatures of an AP monopropellant flame (~ 1400 K) [13] that later react with the fuel-rich decomposition of HTPB in the final flame as shown in Eqs. 2, 3. The relative importance of these three flames depends on the AP grain size and the pressure [5, 13], where with increasing pressure the monopropellant flame dominates as reactions occur faster with less time for the fuel rich species from the binder to diffuse into the decomposed oxidizer. From a measurement point-of-view, these first two flames, the monopropellant flame and primary diffusion flame, occur within 100 μm of the burning surface at 1 atm [4, 5] which has led to several counterflow flame studies of particular AP/HTPB species to examine certain reactions at increased physical scales [16, 17]. This also suggests our measurements occur predominantly within the third and final flame regime, where the highest temperatures are experienced. A cartoon of the three flame zones is shown in Fig. 1b based on figures in Refs. [4, 13].
The propellant studied in this experiment is aluminized, creating additional combustion and diagnostic complexity and very high flame temperatures. The addition of aluminum in solid propellants is typically done to increase the specific impulse of the device and suppress combustion instabilities [6]. As the AP/HTPB material is consumed, these micron to sub-micron aluminum particulate are exposed to the burning surface where the they experience temperatures above their melting point (933 K) and coalesce to form larger 50–200 μm agglomerates [6]. These larger particles are released into the gas-phase plume where they may ignite and take tens of milliseconds to burn while contributing significantly to the plume temperature and heat transfer to the surrounding environment. From an accident scenario point-of-view, understanding the heat transfer from metalized propellant flames is a key safety factor.
Given the low levels of N2 expected in hot propellant gases and our desire to avoid spatial averaging effects associated with entrained air, we turned our focus to the decomposition of the HTPB binder along with products of reactions of HTPB with AP oxidizers (i.e. P2 from Eq. 2) as fruitful sources of molecular hydrogen to probe with a new CARS instrument. By probing the fuel, as opposed to inert nitrogen, the measurements are only possible in fuel-rich plume regions, minimizing any spatial averaging effects. We expanded on our previous demonstration of successful gas-phase measurements in this challenging environment by reporting rotational temperatures from probing the Q-branch Raman transitions of molecular hydrogen by means of a now well-established hybrid fs/ps CARS approach [18,19,20,21,22] during three different propellant burns at 0.82 atm.
Additionally, there are numerous examples of successful H2 CARS thermometry from the nanosecond [23,24,25,26], picosecond [27, 28], and femtosecond regimes [29,30,31,32,33], but there remains discrepancies in the calculation of the H2 Raman frequencies and the dephasing processes of the CARS signal. We include a practical discussion of these complexities and detail how they are mitigated with early probing of the Raman coherence with a short duration probe pulse. Characterization of the accuracy and precision of our resulting CARS instrument and model fitting procedure is performed in canonical near-adiabatic flames while once again demonstrating the effectiveness of time-delayed probing in performing thermometry measurements in the challenging metal-particle laden propellant flames.
2 Theory
Time-domain modeling of hydrogen CARS is found in the literature, with the first example of femtosecond CARS modeling on H2 performed by Lang et al. [29]. We summarize the efforts of others in this section while also highlighting discrepancies in the previous short-pulse H2 CARS literature to bring them to the attention of the combustion diagnostics community. We will additionally show that with the use of a temporally short asymmetric probe pulse at early delays numerous issues with high-fidelity modeling of time-domain H2 CARS are avoided.
2.1 CARS Model
The impulsively driven time-domain CARS model used in this work is based on previous work for pure-rotational CARS [19] and vibrational N2 CARS [34, 35]. By delaying the probe pulse in time from the femtosecond preparation pulses, only the resonant Raman response, \(\chi \left( t \right)\), for H2Q-branch transitions (Δ\(\nu\) = 1, ΔJ = 0) is observed and is modeled using the simple Fourier-type expression:
where the sum is taken over all thermally populated vibrational, v, and rotational, J, levels; \(\omega_{\nu ,J}\) are the Raman frequencies; and ΓJ are the collisional linewidths. The weight terms, Wv,J, account for the ro-vibrational populations, centrifugal distortions, and the Raman cross section and are given by
where Nv,J are the Boltzmann populations of the levels coupled by the Raman transition; ωo is the optical frequency of the probe laser pulse (800 nm); \(\,F_{v,J}\) is the Herman–Wallis factor accounting for the coupling of vibrational and rotational states; and (aʹ)2 and (γʹ)2 are the isotropic component and anisotropy of the polarizability derivative, respectively. For H2, the Raman signal is dominated by the isotropic contribution, with authors often neglecting the anisotropic component [36]. Using the depolarization ratios from James et al. [37], we estimated the relative contributions of both the isotropic and anisotropic components and determined the J-dependent contribution of the anisotropy changed the relative transition strengths by < 2% for J > 1. Similarly, with a near-IR probe pulse of 800 nm, the relative intensity change from the frequency dependence of the Raman cross sections is also negligible. However, for a light diatomic molecule like H2, the centrifugal distortion of a rotating dipole couples the rotational and vibrational motions of the molecule, leading to non-negligible changes in the intensity of spectral lines especially at higher J states. From the guidance of several works by Marrocco [38,39,40], we implemented the J-dependent Herman–Wallis factors from Tipping and Ogilvie [41], assuming the isotropic and anisotropic components are the same. Neglecting these factors would lead to an overestimation in the fitted temperature. For completeness, we include the (v + 1) term in the cross section, but we only consider contributions from the ground vibrational level, v = 0, because the bandwidth of our instrument was insufficient to detect signal from higher vibrational states.
The CARS signal pulse at probe delay, τ, may finally be calculated by
where Epr is the electric-field envelope of the assumed transform-limited probe pulse introduced at delay, τ, from the pump and Stokes pulses. The beating patterns within χ(t) are time-gated by the probe pulse and converted to frequency space via Fourier transform, where the predicted spectral profiles will differ based on uncertain values of the ωv,J.
2.2 Energy levels of hydrogen
This model assumes that all contributions to χ(t) are impulsively prepared by near-transform-limited pump and Stokes pulses and are, therefore, exactly in phase at time t = 0 [35]. As time evolves, these cos(ωv,Jt) contributions oscillate at the Raman frequencies and the specific form of χ(t) is then determined by the relative phase accumulated by each v,J transition and the resulting beat patterns. Modeling of CARS spectra requires highly accurate measurements of the ωv,J. The low molecular weight of hydrogen results in widely spaced rotational and vibrational energy levels, creating issues when calculating their energy term values. Published diatomic constants for hydrogen are quite poor at extrapolating to energy levels outside those used for the determination of the constants themselves [42]. Numerous authors have noted this [29, 43,44,45,46], and opted for an alternative model by means of a double power series originally developed by Dunham [43], with their own set of coefficients to match experimental results [42, 47]. The theory itself has additionally improved with nonadiabatic corrections added to eliminate energy level discrepancies seen between theory and measurements at high rotational levels [48,49,50].
The various reported energy levels lead to different Raman frequencies, which result in different beating patterns in the time-domain Raman coherence calculated via Eq. 4. When these Raman coherences are time-gated by the probe pulse, as per Eq. 6, and converted to frequency space, the spectral profiles will be different. This is illustrated in Fig. 2a, b, where H2 CARS spectra at T = 3000 K are calculated for probe pulse delays of τ = 2.66 ps and τ = 30 ps, using the measured form of Epr from our experiments. Differences between the calculated spectra grow with increased probe delay because of the change in accumulated phase between the cos(ωv,Jt) terms in Eq. 4. Uncertainty in literature values for hydrogen energy levels is illustrated in Fig. 2c–e. Even as low as J = 7, the difference between the Morse-potential-predicted values (such as the value provided by the Sandia CARSFT code [51]) and the measured values from Dabrowski [47] is already 1.3 cm−1 (Fig. 2d) and is readily seen even at the earliest probe delays by a shift on the wavenumber axis (line near 4000 cm−1).

Synthetic CARS spectra for different models of the hydrogen energy levels at probe delays of a 2.66 ps, and b 30 ps for a pressure of 0.82 atm using experimentally determined probe laser pulse shapes. c Example of the discrepancies of H2 Q-branch transition frequencies from experimental data, Morse potential calculations, and a power series approach along with (d) the corresponding residuals with respect to the Dabrowski dataset. The legend in (d) also applies to the data in (c). The hydrogen rotational population distribution at 2000 and 3000 K is shown in (e) for reference
At the small-time delay of 2.66 ps used in our experiments, the accumulated phase is small and so that uncertainty in the ωv,J do not significantly impact the measured CARS spectra. While not physically accurate, one can reasonably correct for this discrepancy by a slight change in the detector pixel-to-wavenumber calibration to match the chosen set of Raman frequencies. However, at later times, such as the τ = 30 ps case shown in Fig. 2b, the spectral intensity profile clearly varies depending on the selection of energy levels. While we encourage the use of a power series expansion approach [45] (i.e. the blue curve in Fig. 2a, b and the values used in our model), or just using the experimental values from Dabrowski [47], (gaps in the Dabrowski data are filled elsewhere [52], but for J levels outside of the thermal scope of this work), this discrepancy in the correct selection of energy levels is mitigated in our experiments by selection of an early time delay of τ = 2.66 ps. Measurements at such short probe delays are enabled by a time-asymmetric probe pulse generated by removing bandwidth from an initially fs pulse using a Fabry Pérot etalon, as well as a flexible frequency axis calibration.
Hydrogen energy levels have a nonlinear dependence with density as well, which are small (order of ~ 10−3 cm−1) compared to the ~ 1 cm−1 energy level deviations discussed above at the density of ~ 0.07 amagat in our high-temperature propellant flame measurements near 3000 K. This density dependence can be addressed with shift coefficients to correct the Raman frequencies. These correction factors were determined both by frequency-domain [53] and time-domain [54] measurements, with a power-series expression of the form
where \(\omega_{J}^{0}\) is the transition at zero density, \(\rho\) is the density in amagat (amagat = \((P/P_{{\text{o}}} )(T_{{\text{o}}} /T)\), where \(P_{{\text{o}}} = 101.325\; {\text{kPa}}\) and \(T_{{\text{o}}} = 273.15 \;{\text{K}}\)), and \(\alpha_{J}\) (in units of 10−3 cm−1/amagat) and \(\beta_{J}\) (10−6 cm−1/amagat) are the fit coefficients. Only the linear shift coefficients, \(\alpha_{J}\), are considered here, as any higher corrections are insignificant for this work. Rahn et al. [53] tabulate the linear shift coefficients for temperatures from 295 to 1000 K for J = 0–5, which we linearly extrapolated to our flame temperatures.
2.3 H2 linewidths: Doppler and collisional dephasing
Due to its low molecular weight, hydrogen experiences unique dephasing characteristics with increasing density [55]:
- 1.
At low densities, the thermal motion of the molecules broadens the linewidths (Doppler broadening) and increases coherence dephasing rates. The dephasing is a Gaussian decay in the time domain, leading to Gaussian lineshapes in the spectrum.
- 2.
With increasing density, velocity changing collisions act to decrease the linewidths and slow coherence dephasing rates, which is known as the Dicke effect [56]. This process becomes important when the wavelength of radiation becomes comparable to the mean-free path [57] and has generally been modeled as a Lorentzian lineshape with a corresponding exponential time-domain decay.
- 3.
At even higher densities, elastic and inelastic collisions broaden the linewidths and increase dephasing rates. The decay is exponential, leading to Lorentzian lineshapes.
It is common in the literature [23, 25, 27, 29,30,31] to express this change in linewidth with density as a summation of the velocity changing and phase changing collisions into one effective linewidth, \(\Gamma_{J}\), defined as
where \(D_{{\text{o}}} = 0.01176T^{0.8314}\) cm2-amagat/s is the assumed rotational-state-independent optical diffusion coefficient [58] for hydrogen; c is the speed of light; \(\rho\) is the density in amagat; and \(\gamma_{J}\) is the rotational-state-dependent collisional linewidth. This transition through different linewidth and dephasing regimes of the H2 Q-branch has been examined in the frequency domain (see [59], Fig. 2) and in the time domain (see [27], Fig. 2 or see [60], Fig. 3) as a function of density.

a Linewidths of the H2 Q(1) Raman transition, as a function of density at 300 K, from various sources and from the models discussed above. The values from De Martini et al. are forward scattered linewidths, producing a minimum measured Doppler linewidth for the transition based on the collection angle. The Doppler limit is expressed as a dashed line, above which linewidth values from Eq. 8 are no longer physical. The onset of motional narrowing is marked by the density at which the mean free path, \(\lambda_{{{\text{mfp}}}}\), is equal to the wavelength of radiation, \(\lambda_{Q\left( 1 \right)}\), as expressed by Murray et al. [57]. b An extrapolation of the models to the mean temperature expected in the propellant flames, along with the operating regime of the propellant burn in Albuquerque, NM (P = 0.82 atm, T range from 2000 to 3000 K)
However, this type of formulation assumes only an exponential decay of the Raman coherence (and Lorentzian spectral lineshapes) and would overpredict the dephasing in the Doppler regime [23]. Another, more complete, approach in the low-density regime is to separately consider the Doppler and collisional effects by assuming the translational (Doppler) and rotational (collisional) dephasing are statistically independent such that the dephasing term, \(H\left( t \right)\), becomes [61]
where \(H_{{{\text{ph}}}} (t) = \exp \left( { - \Gamma_{J} t} \right)\) is the collisional dephasing exponential decay from Eq. 4, and \(H_{v} \left( t \right)\) is a Doppler dephasing term defined as
Doppler dephasing is a function of the transition wavenumber, \(k_{{\text{o}}} = 2\pi \omega_{J}\), the dispersion of thermal velocities, \(\sigma_{v} = \sqrt {k_{{\text{B}}} {T \mathord{\left/ {\vphantom {T m}} \right. \kern-\nulldelimiterspace} m}}\), where m is the molecular mass, and the velocity correlation time, \(\tau_{v} = {{D_{{\text{o}}} } \mathord{\left/ {\vphantom {{D_{{\text{o}}} } {\left( {\rho \sigma_{v}^{2} } \right)}}} \right. \kern-\nulldelimiterspace} {\left( {\rho \sigma_{v}^{2} } \right)}}\). This method separates the Gaussian dephasing of Doppler broadening from the exponential dephasing of collisional broadening [62] and has been verified experimentally on pure-rotational Raman spectra of H2 [55, 61].
For this experiment, where flame temperatures reach ~ 3000 K at the local atmospheric pressure of 0.82 atm, the expected densities are as low as ~ 0.07 amagat—well within the Doppler broadened regime. Figure 3a shows results from the dephasing measurements of the Q(1) line of H2 as a function of density, by both high-resolution Raman measurements in the frequency [59, 63, 64] and time domains [27, 60], in comparison to the functional forms of the broadening mechanisms presented earlier in Eqs. 8 and 10. Note that forward [59] and backward [65] Raman scattering measurements produce significantly different linewidths in the Doppler regime [64], as they become collection-angle dependent [57]. Our implementation of BOXCARS phase matching is closely approximated by forward scattering, as are all of the results reproduced from the literature in Fig. 3.
As seen in Fig. 3a, the three distinct broadening regimes are evident, with most of the experimental effort in the Dicke narrowing regime. However, for lower densities, the model from Eq. 8 deviates significantly from the Doppler limit (Eq. 10), resulting in a large overestimation of the linewidth [23]. Therefore, use of Eq. 8 has no physical meaning for densities where the predicted linewidths are larger than the Doppler limit, defined by the dashed line in Fig. 3. This is further evidenced by the forward scattering results from De Martini et al., showing a significant change in the measured linewidth for the lowest densities examined compared to what Eq. 8 predicts. Extrapolating these equations for the mean temperature experienced in these burns (T = 2500 K) in Fig. 3b, while uncertain, shows that our propellant burn conditions are well within the Doppler regime, encouraging the use of Eq. 10 over the more commonly used Eq. 8.
2.4 Mitigation effort: etalon probe pulse
By utilizing a temporally short probe pulse at small delays, such as the time-asymmetric pulse generated from a Fabry–Pérot etalon [66] or Gaussian pulse generated via second-harmonic bandwidth compression (SHBC) [67], the decay of the Raman coherence is negligible in atmospheric pressure flames. We chose a probe pulse shaped by a Fabry–Pérot etalon, as the temporally sharp leading edge allows us to position the probe pulse close to the preparation pulses in time without sampling the nonresonant CARS background, and the temporal width only gates a small portion of the Raman coherence, as seen in comparison to a SHBC probe in Fig. 4a. This allows us the significant advantage of neglecting the finer details of both dephasing processes and energy level discrepancies in our molecular coherence model. Such a temporally short probe pulse necessarily exhibits bandwidths of a few cm−1, thus limiting the spectral resolution of the CARS instrument. As seen in the synthetic spectral profiles in Fig. 4b, energy level spacings for hydrogen are large enough to prevent this from being as much of an issue as it would be for heavier molecules such as in N2 Q-branch thermometry [34]. An additional drawback is the limited pulse energy one can achieve through an etalon (for us only 15 μJ), which in turn will raise the detection limit and lower the signal-to-noise ratio in our CARS spectra.

a Example synthetic coherence with the probe electric field profile and b model spectra for different temperatures with the etalon-generated pulse at a probe delay of 2.66 ps
The time-dependent electric field amplitude of the time-asymmetric probe pulse is compared to an SHBC-generated Gaussian pulse and the calculated Raman coherence in hydrogen at T = 3000 K. Both probe pulses are achievable in our lab, but the Gaussian temporal profile of the SHBC would lead to significant nonresonant background generation, contaminating the Raman-resonant signal for τ = 2.66 ps delay of the probe pulse used in our experiments. Calculated spectra using the time-asymmetric etalon pulse for temperatures of T = 1000, 2000, and 3000 K are shown in Fig. 4b to demonstrate the temperature dependence of the H2 CARS signal. Experimental spectra are fit for temperature to libraries of precomputed synthetic spectra of this type for a given probe temporal profile and probe pulse delay.
3 Experimental setup
3.1 Optical setup
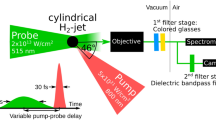
The optical setup for the hybrid fs/ps CARS system is shown in Fig. 5. As seen in the energy level diagram on the left side of Fig. 5, broadband fs pump and Stokes pulses arrive simultaneously at the measurement volume at t = 0 ps, exciting a Raman coherence in H2, which is probed with the frequency narrow pulse at a short delay, t = τ. The sharp leading edge of the probe pulse in time allows for short probe delays, eliminating any nonresonant background by avoiding the fast-decaying electron response to the initial preparation pulses and negating the need of a dephasing model for the Raman coherence. Delays for the experiments presented in this work were ~ 2.6 ps, achieved with automated stages.

Optical setup used for the H2 CARS thermometry, including insets of the energy level diagram, the phase matching, and the measurement length on both a FWHM and a 5–95% basis
A Ti:sapphire regenerative amplifier (Spectra-Physics Spitfire Ace, 800 nm, 50 fs, 1 kHz, 11.3 mJ) provides pulse energy for pump, Stokes, and probe beams in the CARS process. The three laser beams are arranged in a folded BOXCARS phase matching scheme [68] at the f = 300-mm beam crossing lens seen as an insert on the bottom right of Fig. 5. A ~ 50-fs CARS pump beam at 150 μJ/pulse was obtained by regulating 1.5 mJ through a neutral density filter with 10× attenuation. A Stokes-shifted beam at ~ 4100 cm−1 from the pump frequency prepared the Q-branch of molecular hydrogen and was produced by down-converting 4 mJ of 800-nm radiation in a commercial optical parametric amplifier (OPA, Light Conversion TOPAS), with ~ 80 μJ of 1200-nm Stokes pulse energy delivered to the measurement volume. These preparation pulse energies are relatively high for 50 fs pulses, but lowering the pulse energies did not cause any significant changes to the spectrum, as might occur as a result of stimulated Raman pumping and/or semi-permanent alignment of molecules, providing us confidence to proceed with the propellant burns at these elevated energies.
A frequency narrow, time asymmetric, picosecond-duration probe pulse was generated by passing 1.5 mJ of 800-nm radiation through an angle-tuned, air-spaced etalon (TechOptics, free spectral range = 30.27 nm, finesse = ~ 104 at 800 nm) to achieve an exponentially decaying pulse shape with ~ 1.5 ps decay time, ~ 5.3 cm−1 FWHM spectral bandwidth and 15 μJ probe pulse energy, as shown in Fig. 4. The measurement length in the beam-propagation direction was determined by scanning a 140-μm glass coverslip through the measurement volume with the three beams overlapped in time and attenuated by upstream neutral density filters. The nonresonant signal generation from the coverslip was captured on the detector and integrated for each spatial location of the coverslip. The resulting measurement length of ~ 2.1 mm incorporates 90% of the total signal level as shown in the bottom left of Fig. 5.
Each of the pump, Stokes, and probe beam lines featured a half waveplate to obtain parallel polarizations at the beam crossing and maximize the CARS signal generated near 600 nm. Telescoping lenses were inserted in the Stokes and probe beam lines to match the foci of these beams to the 800-nm pump. The pump and probe beams shared the same frequency in this experiment, which necessitated the selection of a 1200-nm Stokes pulse wavelength to yield a CARS signal near 600-nm, which can be spectrally isolated from the input laser pulses [69, 70]. This scheme was preferable to a 600-nm pump, 800-nm Stokes pair, which would have resulted in a CARS signal that was frequency degenerate with the pump [18, 20, 22, 34], resulting in significant contamination of the measured spectra by intense scattering of pump radiation from metal-based particulate in the propellant flame. The resulting 600-nm CARS radiation was collimated with an f = 400-mm lens and dispersed with a 1-m spectrograph with 1800 l/mm diffraction grating, where a 100-μm slit results in a spectral resolution of 1 cm−1 when detected using an Andor Newton electron-multiplying CCD operating in full-vertical-bin mode. Horizontal binning (i.e. spectral binning) of 4 pixels increased CARS signal strength and resulted in a spectral dispersion of ~ 0.7 cm−1 per superpixel. The detector was triggered by the fs amplifier to acquire single-laser-shot spectra at the 1-kHz of the femtosecond amplifier.
3.2 Measurements in a Hencken burner
In order to test the accuracy and precision of our hydrogen thermometry instrument, we first measured the temperature in near-adiabatic fuel-rich hydrogen-air flames stabilized on a Hencken burner [19, 24]. Flowmeter limitations restricted conditions to an equivalence ratio range of 1.0–1.8, corresponding to adiabatic equilibrium temperatures of 2132–2379 K and H2 concentrations from 1.5 to 20.8%. Measurements were recorded approximately 25 mm off the burner surface at the center of the 50 mm square burner exit. Measured resonant spectra were background subtracted by acquisitions with the probe beam blocked and normalized by the nonresonant spectrum recorded in room air. These corrected spectra were fit to a temperature library of synthetic spectra generated with the experimental probe profile at the delay used for the measurements.
3.3 Propellant composition and burn operation
The three propellants examined in this work were all the same composition of approximately 70% AP, 10% HTPB, and 20% aluminum by weight. These were formed into 7-mm diameter strands approximately 53 mm long and molded into points at the top where resistive heating elements were mounted and used to ignite the propellant. An epoxy coating was added to the sides and bottom of the strand, encouraging the burn to proceed downward with a relatively flat burning surface. The propellant burn rates, estimated by the physical length of the strand and the time from ignition to extinction, was on average 1.49 mm/s for the three burns.
A typical propellant burn procedure is shown in Fig. 6 from the ignition phase through to the end of the burn. Initially, a glass coverslip is placed at the beam crossing and the propellant stick is placed directly under it by means of a three-axis translation stage. The coverslip marks the center of the measurement probe volume once the maximum nonresonant CARS generation is found by translating the glass along the propagation direction of the CARS laser beams. The glass coverslip is removed, and the stick is translated up into the beam crossing such that the beams are hitting the propellant strand. Ignition of the propellant by the beams themselves was prevented by having an upstream neutral density filter which was removed right before the propellant was ignited by a resistive heating element attached to the tip of the propellant strand; the orange glow of the heating element can be seen in the upper left in Fig. 6. The camera acquisition is started before the propellant is ignited, thus the early frames are mostly all background noise from the detector. The beam crossing remains stationary, while the propellant burns down, thus measurements later in time correspond to positions farther from the burning surface and higher in the propellant flame plume.

Example timeline of the propellant burn. Each time segment shows a digital photograph of the propellant (first row), an example spectrum acquired during that phase (second row), and a cartoon of events (third row)
Once the propellant is lit, there is a short time when the propellant is burning but the beams remain blocked by the unburned propellant strand. During this phase, there is non-negligible broadband emission from the luminescent burn visible on the detector. This emission is highly dynamic, but relatively flat, so that background corrections to the single-laser-shot spectra were performed using a simple quadratic polynomial fit to the intensity values at select reference pixels.
Eventually, the stick burns low enough such that the beam crossing is now in the multiphase plume just above the burning surface. At these early times, colder hydrogen gas is probed, evident by the relative weighting of the Raman lines with a bias towards higher wavenumbers. This suggests these early measurements, closest to the burning surface, are measuring gases near the monopropellant and primary flames where temperatures are colder [4] before acquiring spectra later in time and physically farther away from the burning surface in the final flame.
Shortly after the first detected resonant CARS signal, the CARS spectrum stabilized around a higher temperature, evidenced by the larger weighting towards lower wavenumbers. Spectra of reasonable SNR are observed for up to ~ 10 s, suggesting a high concentration of higher temperature hydrogen that tends to persist in the plume. Finally, at late times, the propellant has burned down low enough such that there is no remaining measurable hydrogen in the CARS measurement probe volume. The detector sees a slightly elevated background from the flame emission, although not as intense as early in the burn.
4 Results and discussion
4.1 Hencken burner results
Results for equivalence ratios of 1–1.8 are shown in Fig. 7. Lower equivalence ratios featured inadequate concentrations of hydrogen, coinciding with poor signal-to-noise, and thus no spectra were obtainable. Our results are compared to adiabatic equilibrium predictions [71], where on average we consistently measure temperatures 5–6% lower than the adiabatic equilibrium value when temperatures were derived from fits to CARS spectra averaged for 100 laser shots. This low-temperature bias may be from the adiabatic-equilibrium assumption in the calculated temperatures as the available gas flow rates for operation of our Hencken burner at fuel-rich conditions were limited such that the diffusion flamelets were not adequately lifted off the burner surface, forcing short run times in practice and potentially lowering the flame temperature.

a Results of spectral fitting of single-shots and 100-shot averages of data recorded in a near-adiabatic Hencken flame for different equivalence ratios. b Precision of the temperature fits as a function of SNR in the Hencken burner (top) and the measured SNR during the first 2 s (2000 shots) for the three propellant burns (bottom)
Mean temperatures obtained from fits to 2000 single-laser-shot spectra are compared against those from fits to 100-shot-averaged spectra to demonstrate the signal to noise ratio (SNR) dependence of our H2 CARS instrument. Here \({\text{SNR}} = {{N_{{{\text{e}}^{ - } }} } \mathord{\left/ {\vphantom {{N_{{{\text{e}}^{ - } }} } {\sqrt {R^{2} + N_{{{\text{e}}^{ - } }} } }}} \right. \kern-\nulldelimiterspace} {\sqrt {R^{2} + N_{{{\text{e}}^{ - } }} } }}\), where \(N_{{{\text{e}}^{ - } }}\) is the peak photoelectrons on the detector and R is the rms of the detector readout noise (9.4 e− for our detector). With increasing fuel concentration, the SNR also increases; thus the temperatures obtained from the mean of 2000 single-laser-shot measurements and from fits to 100-shot-averaged spectra converge to the same values in Fig. 7a for equivalence ratios greater than about 1.4. As seen in Fig. 7b, the precision of our single-laser-shot temperature measurements, represented by a single standard deviation relative to the mean, approaches ~ 3% as the signal to noise increases, but never reaches the 1–1.5% precision floor experienced with a previous pure-rotational CARS instrument from our lab [19]. We attribute the decreased precision of this rovibrational instrument to the use of an OPA to generate the Stokes pulse. The quality of the nonresonant spectrum measured in room air changed significantly throughout the day, only to be recovered by manipulation of the OPA optics. The SNR during a propellant burn decayed with time, with an average SNR of 45 during the first two seconds of each burn, corresponding to a precision of ~ 6%. Histograms of the measured SNR during the first two seconds for each of the three propellant burns examined in this paper are shown in the bottom of Fig. 7b.
4.2 Propellant burn results
Applying this instrument to the propellant burns was enabled by the large concentration of H2 from the decomposition of HTPB. Representative fits to single-laser-shot spectra from the propellant flames are shown in Fig. 8, with results displayed over a range of temperatures to demonstrate the good-quality SNR on single shots even at temperatures near 3000 K. Some of the best high- and low-temperature fits are featured in the first row, while more typical fits are shown in the bottom row of Fig. 8.

Some of the best (top) and typical (bottom) fits to single-laser-shot experimental spectra acquired in the propellant flames
The time history for all single-laser-shot temperatures and the corresponding running average for every 100 retained laser shots for each propellant burn is shown in Fig. 9. The approximate height above the burning surface, based on the observed burn rate of 1.49 mm/s, is also indicated. Within the first 500 ms of burns 1 and 3 (approximately up to 400 μm above the burning surface), the average measured temperature rises from ~ 2100 K to over ~ 2500 K. The second burn does not feature this rise in temperature, most likely due to the relative heights of the beams hitting the sticks before ignition. The top of the sticks were molded into points; thus it is possible that for the second burn the beams were hitting the top portion of the point, and thus directly after ignition the smaller volume of material quickly burned leaving the beams physically higher above the burning surface than in the other burns.

Plots of the temperature as a function of burn time for the 3 burns. The dots represent all the single-laser-shot temperature results, while the lines represent a running average of temperatures obtained from fits to every 100 spectra retained for fitting. All temperatures are also shown as the approximate height above the burning surface based on the measured burn rate of 1.49 mm/s
The height of the final flame in AP propellants, where the highest temperatures are expected, is typically predicted to be within 100–500 μm of the burning surface [5]. Therefore, based on our procedure, burn rate, and laser repetition rate, the first 67–335 acquired spectra may be within the monopropellant and primary diffusion flame regime closest to the propellant burning surface. While H2 itself does not contribute to the monopropellant flame (predominately NH3 + HClO4 decomposition products), the high temperatures, ~ 1400 K [13], from the monopropellant flame assist the pyrolysis of the binder into hydrocarbons and H2 [15]. The decomposed H2 will have thermally populated rovibrational levels from proximity to the monopropellant flame, along with elevated temperature H2 products from hydrocarbons reacting with the AP oxidizers in the primary diffusion flame. These species will be relatively cool compared to the temperatures in the final flame farther from the burning surface and may be the source of the colder H2 we measure at these early times.
In the final flame region of the plume, all three burns on average remain at a constant temperature near 2500 K over the remaining ~ 9 s, suggesting a relatively consistent burn on average juxtaposed to the rather violent emission of aluminum particles one sees by eye. The scatter in the fits exceeds the expected precision of our instrument of ~ 6%, or 150 K from a mean of 2500 K, based on the SNR of the burn data for the first 2 s. Therefore, the broad range of fitted temperatures, from 2000 K to over 3000 K, reveal physical temperature fluctuations. Our measurements on average are ~ 600 K below the predicted adiabatic Al/AP/HTPB/Air flame temperature of ~ 3100 K for an equivalence ratio of 1.0 [10, 71], but approach that value on single laser shots.
Histograms of fitted temperatures from the three different burns are detailed in Fig. 10, along with the corresponding nonresonant CARS spectra acquired in room air. In total, the three burns occurred over the course of ~ 2 h, leading to a significant drift in the nonresonant spectrum in time. As discussed previously, we attribute this change of the nonresonant spectrum to the behavior of the Stokes pulse derived from the OPA. To mitigate these issues, nonresonant spectra were recorded in room air just before and directly after each burn to bound the Raman-resonant H2 CARS spectra with their corresponding normalization curves that account for the finite bandwidth of the CARS instrument. All three burns led to near-identical before and after nonresonant spectra, and thus the average of the two was used to normalize the resonant spectra for each burn. Given the different correction factors of the nonresonant spectra for each burn, but the consistent fitted temperatures, this gives us significant confidence in our fitting procedure and the measured temperatures shown in Fig. 9 demonstrate the consistency of these propellant burns.

a Histograms of fitted temperatures of the three burns and b the corresponding nonresonant spectrum taken before and after each burn
The mean values of the three burns only differ by 29 K, with a mean temperature of 2494 K encompassing all three burns. The data retention rate, defined by upper limits placed on the residuals to the model fits, was only ~ 32% on average for the three runs over the first 10 s of the burn, but as high as ~ 57% on average for the first second, where the measurements are made closest to the propellant surface. At early times, dynamic background emission plagued the retention rate, while the low hydrogen concentration at late times (i.e. physically farther away from the burning surface) limited signal-to-noise and prevented high retention. Further time gating of the resonant CARS signal on the detector, such as with a high repetition rate liquid crystal shutter, would help mitigate the intense flame luminosity at early times and is a source of improvement for future work.
Figure 11 compares the temperature results from burn 1 of this work to other measurements of temperature from our lab on the same aluminized AP propellant material using two-color pyrometry of aluminum particulate [9] and pure-rotational CARS of N2 and O2 [10]. The rotational CARS measurements exhibit appreciable low-temperature content compared to the aluminum pyrometry results and the H2 CARS results, indicative of the fact the rotational CARS measurements were not only generally confined to the flame plume, but also captured the (cold) ambient gases surrounding and mixing with the flame. However, the highest measured temperatures of the rotational CARS instrument are up to ~ 3200 K which agrees well with the H2 CARS results, demonstrating the ability of the rotational CARS instrument to capture the full range of temperatures from the hot plume to the cold surrounding ambient air. The aluminum pyrometry measurements, recorded within millimeters of the burning surface, show a similar distribution to the H2 CARS measurements with a mean temperature only 5 K higher at 2488 K. This suggests the aluminum particles are very similar in temperature to the gas phase hydrogen, or possibly that the pyrometer may have a gas-phase bias.

Comparison of the different temperature measurements from our lab in an AP/HTPB/AL propellant burn. The pyrometry is from a 2-color pyrometer measuring the solid aluminum temperatures, H2 CARS is the gas phase rotational temperature of H2 (burn 1 from this experiment), and O2, N2 rotational CARS is the gas phase rotational temperature of N2. Each histogram also shows an outline of the other two temperature distributions for ease of comparison
The summation of these gas- and solid-phase temperature measurements provides useful model validation data near the burning surface and establishes a baseline for the expected plume temperature from these propellants. While the averaged H2 results suggest a consistent burn temperature, the measurement lengths of 1.8 and 2.1 mm used for the pure-rotational [10] and H2 CARS measurements, respectively, preclude direct comparisons of the large thermal gradients shown in detailed models of the burning surface. This provides avenues forward for additional study, including increasing the dimensionality of the CARS measurements to better examine thermal gradients near the burning surface using one-dimensional “line CARS” approaches demonstrated recently [72] and coupling these findings to heat transfer models to understand the impact of propellant fires on the surrounding environment.
5 Summary and conclusions
A hybrid fs/ps hydrogen CARS instrument was used to measure the plume temperatures above the burning surface of an aluminized propellant strand at local atmospheric pressure (0.82 atm). The probe beam was a time-asymmetric exponentially decaying pulse obtained by removing bandwidth from an initially femtosecond pulse using an air-spaced etalon. The short, 1.5-ps duration and asymmetry of the probe pulse enabled us to obtain spectra during the earliest portions of the H2 Raman coherence at a probe time delay of 2.66 ps. This small probe delay coupled with a short probe duration enabled accurate thermometry despite uncertainty in the combined Doppler and collisional dephasing models and in the hydrogen energy levels used in our time-domain CARS model. When applied to H2-air near-adiabatic flames stabilized on a Hencken burner, this instrument demonstrated a precision approaching 3–4% for high signal-to-noise spectra, limited mostly by the quality of the Stokes pulse generate by an OPA. The accuracy, compared to adiabatic equilibrium predictions, was near 5% for equivalence ratios from 1 to 1.8 for average spectra.
The CARS measurement probe volume was stationary and as the propellant burned down, measurements were recorded at increasing heights above the burning surface in the metal-particle-laden plume. While some intense broadband flame emission was recorded simultaneously with the CARS signal, high-quality resonant spectra were recorded as late as 10 s into each burn, corresponding to measurements approximately 15 mm above the burning surface. At early times, close to the burning surface, the measured temperature was approximately 2100 K, followed by a rise to an average temperature of just over 2500 K for the remainder of the burn. The average temperature from three burns differed only by 29 K and in general compares favorably to previous measurements of temperature from solid-phase aluminum pyrometry. In addition, the gas phase thermometry provided by hydrogen CARS limits the temperature measurement to the hot plume gases unlike our previous rotational CARS measurements which, while capturing the full range of gas temperatures from the hot plume to the cold ambient air, was subject to spatial averaging effects that limit our ability to measure peak flame temperatures. These results also provide further evidence of the fitness of these hybrid CARS instruments in probing multiphase environments.
References
E.H. Richardson, J.M. Blackwood, M.J. Hays, T. Skinner, Solid rocket launch vehicle explosion environments, NASA Technical Report M15-4253 (2014)
M.W. Beckstead, K. Puduppakkam, P. Thakre, V. Yang, Prog. Energy Combust. Sci. 33, 497 (2007)
T.L. Jackson, AIAA J. 50, 993 (2012)
C. Dennis, B. Bojko, Fuel 254, 115646 (2019)
T. Edwards, Solid propellant flame spectroscopy, AFAL-TR-88-076 (1988)
K.K. Kuo, Fundamentals of Solid-Propellant Combustion (AIAA, 1984)
P. Bucher, R.A. Yetter, F.L. Dryer, T.P. Parr, Combustion 27, 2421 (1998)
D.R. Guildenbecher, M.A. Cooper, W. Gill, H.L. Stauffacher, M.S. Oliver, T.W. Grasser, Opt. Lett. 39, 5126 (2014)
Y. Chen, D.R. Guildenbecher, K.N.G. Hoffmeister, M.A. Cooper, H.L. Stauffacher, M.S. Oliver, E.B. Washburn, Combust. Flame 182, 225 (2017)
S.P. Kearney, D.R. Guildenbecher, Appl. Opt. 55, 4958 (2016)
M. O’Neil, N.A. Niemiec, A.R. Demko, E.L. Petersen, W.D. Kulatilaka, Appl. Opt. 57, 1910 (2018)
E.J. Beiting, Appl. Opt. 24, 3010 (1985)
M.W. Beckstead, R.L. Derr, C.F. Price, AIAA J. 8, 2200 (1970)
O.P. Korobeinichev, N.E. Ermolin, A.A. Chernov, I.D. Emel, Combust. Explos. Shock Waves 28, 366 (1992)
M. Jeppson, M. Beckstead, Q. Jing, in 36th AIAA Aerosp. Sci. Meet. Exhib. (1997)
T.P. Parr, D.M. Hanson-Parr, M.D. Smooke, R.A. Yetter, Proc. Combust. Inst. 29, 2881 (2002)
T.P. Parr, D.M. Hanson-Parr, M.D. Smooke, R.A. Yetter, Proc. Combust. Inst. 30(II), 2113 (2005)
J.D. Miller, M.N. Slipchenko, T.R. Meyer, H.U. Stauffer, J.R. Gord, Opt. Lett. 35, 2430 (2010)
S.P. Kearney, Combust. Flame 162, 1748 (2014)
C.E. Dedic, T.R. Meyer, J.B. Michael, Optica 4, 563 (2017)
A. Bohlin, C. Jainski, B.D. Patterson, A. Dreizler, C.J. Kliewer, Proc. Combust. Inst. 36, 4557 (2017)
D.R. Richardson, H.U. Stauffer, S. Roy, J.R. Gord, Appl. Opt. 56, E37 (2017)
V. Bergmann, W. Stricker, Appl. Phys. B Laser Opt. 61, 49 (1995)
R.D. Hancock, K.E. Bertagnolli, R.P. Lucht, Combust. Flame 109, 323 (1997)
J. Hussong, W. Stricker, X. Bruet, P. Joubert, J. Bonamy, D. Robert, X. Michaut, T. Gabard, H. Berger, Appl. Phys. B Lasers Opt. 70, 447 (2000)
V. Kornas, V. Schulz-von der Gathen, T. Bornemann, H.F. Döbele, G. Prosz, Plasma Chem. Plasma Process. 11, 171 (1991)
W.D. Kulatilaka, P.S. Hsu, H.U. Stauffer, J.R. Gord, S. Roy, Appl. Phys. Lett. 97, 198 (2010)
H.U. Stauffer, W.D. Kulatilaka, P.S. Hsu, J.R. Gord, S. Roy, Appl. Opt. 50, A38 (2011)
T. Lang, K.L. Kompa, M. Motzkus, Chem. Phys. Lett. 310, 65 (1999)
H. Skenderovic, T. Buckup, W. Wohlleben, M. Motzkus, J. Raman Spectrosc. 33, 866 (2002)
T.L. Courtney, A. Bohlin, B.D. Patterson, C.J. Kliewer, J. Chem. Phys. 146, 224202 (2017)
T. Lang, M. Motzkus, JOSA B 19, 340 (2002)
A. Bohlin, C.J. Kliewer, Appl. Phys. Lett. 105, 161111 (2014)
K.N. Gabet Hoffmeister, D.R. Guildenbecher, S.P. Kearney, in 54th Aerosp. Sci. Meet. (2016), pp. 1–13
R.P. Lucht, S. Roy, T.R. Meyer, J.R. Gord, T.R. Meyer, Appl. Phys. Lett. 89, 251112 (2006)
R.L. Farrow, G.O. Sitz, J. Opt. Soc. Am. B 6, 865 (1989)
T.M. James, M. Schlösser, S. Fischer, M. Sturm, B. Bornschein, R.J. Lewis, H.H. Telle, J. Raman Spectrosc. 44, 857 (2013)
M. Marrocco, J. Raman Spectrosc. 41, 870 (2010)
M. Marrocco, Chem. Phys. Lett. 442, 224 (2007)
M. Marrocco, J. Raman Spectrosc. 43, 621 (2012)
R.H. Tipping, J.F. Ogilvie, J. Raman Spectrosc. 15, 38 (1984)
D.K. Veirs, G.M. Rosenblatt, J. Mol. Spectrosc. 121, 401 (1987)
J.L. Dunham, Phys. Rev. 41, 721 (1932)
U. Fink, T.A. Wiggins, D.H. Rank, J. Mol. Spectrosc. 18, 384 (1965)
A. Popovas, U.G. Jorgensen, Astron. Astrophys. 595, 1 (2016)
A. Irwin, Astron. Astrophys. 182, 348 (1987)
I. Dabrowski, Can. J. Phys. 62, 1639 (1984)
W. Kolos, L. Wolniewicz, J. Mol. Spectrosc. 54, 303 (1975)
C. Schwartz, R.J. Le Roy, J. Mol. Spectrosc. 121, 420 (1987)
L. Wolniewicz, J. Chem. Phys. 103, 1792 (1995)
R.E. Palmer, The CARSFT computer code calculating coherent anti-Stokes Raman spectra: User and programmer information, SAND-89-8206 (1989)
H. Abgrall, E. Roueff, F. Launay, J.Y. Roncin, J.-L. Subtil, J. Mol. Spectrosc. 157, 512 (1993)
L.A. Rahn, G.J. Rosasco, Phys. Rev. A 41, 3698 (1990)
T. Lang, M. Motzkus, J. Raman Spectrosc. 31, 65 (2000)
I.G. Konovalov, V.B. Morozov, V.G. Tunkin, A.V. Mikheev, J. Mol. Struct. 348, 41 (1995)
R.H. Dicke, Phys. Rev. 105, 472 (1953)
J.R. Murray, A. Javan, J. Mol. Spectrosc. 42, 1 (1972)
L.A. Rahn, R.L. Farrow, G.J. Rosasco, Phys. Rev. A 43, 6075 (1991)
F. De Martini, F. Simoni, E. Santamato, Opt. Commun. 9, 176 (1973)
S.A. Magnitskiĭ, V.G. Tunkin, Sov. J. Quantum Electron. 11, 1218 (1981)
V.G. Arakcheev, D.V. Jakovlev, V.B. Morozov, A.N. Olenin, V.G. Tunkin, J. Raman Spectrosc. 34, 977 (2003)
L. Galatry, Phys. Rev. 122, 1218 (1961)
M.A. Henesian, L. Kulevskii, R.L. Byer, R.L. Herbst, Opt. Commun. 18, 225 (1976)
J.R. Murray, A. Javan, J. Mol. Spectrosc. 29, 502 (1969)
P. Lallemand, P. Simova, G. Bret, Phys. Rev. Lett. 17, 1239 (1966)
H.U. Stauffer, J.D. Miller, S. Roy, J.R. Gord, T.R. Meyer, J. Chem. Phys. 136, 111101 (2012)
S.P. Kearney, D.J. Scoglietti, Opt. Lett. 38, 833 (2013)
A.C. Eckbreth, Appl. Phys. Lett. 32, 421 (1978)
W.D. Kulatilaka, H.U. Stauffer, J.R. Gord, S. Roy, Opt. Lett. 36, 4182 (2011)
L.M. Thomas, A. Satija, R.P. Lucht, Appl. Opt. 56, 8797 (2017)
B.J. McBride, S. Gordon, Computer program for calculation of complex chemical equilibrium compositions and applications. Part 1: Analysis., NASA Ref. Publ. 1311 (1996)
A. Bohlin, M. Mann, B.D. Patterson, A. Dreizler, C.J. Kliewer, Proc. Combust. Inst. 35, 3723 (2015)
Acknowledgements
The authors would like to thank Yi Chen and Daniel Guildenbecher for providing the aluminum particle pyrometry data, as well as Howard L. Stauffacher, Sam M. Reardon, and Glen White for their expertise in setting up and igniting the propellant sticks. This paper describes objective technical results and analysis. Any subjective views or opinions that might be expressed in the paper do not necessarily represent the views of the U.S. Department of Energy or the United States Government. Sandia National Laboratories is a multimission laboratory managed and operated by National Technology & Engineering Solutions of Sandia, LLC, a wholly owned subsidiary of Honeywell International Inc., for the U.S. Department of Energy’s National Nuclear Security Administration under contract DE-NA0003525.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Retter, J.E., Richardson, D.R. & Kearney, S.P. Rotational hydrogen thermometry by hybrid fs/ps coherent anti-Stokes Raman scattering in the plume of a burning metalized propellant. Appl. Phys. B 126, 83 (2020). https://doi.org/10.1007/s00340-020-07434-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00340-020-07434-3