
Abstract
The manganates Sm1.5–xSr1 + xBa0.5Mn2O7 for x = 0, 0.1, and 0.3 were successfully synthesized by the co-precipitation route. The compounds’ crystalline structures were analyzed through powder X-ray diffraction using the refinement Rietveld method. The refined structure parameters are entirely in agreement with those expected in the case of An + 1BnO3n + 1 for n = 2 corresponding to the iso-structural Sr3Ti2O7 type. Magnetic measurements of Sm1.5–xSr1 + xBa0.5Mn2O7 (x = 0, 0.1, and 0.3) revealed the existence of a predominant antiferromagnetic ordering. Furthermore, isothermal magnetization shows that magnetization saturation is not fully attained at high fields, which can be explained by a strong super-exchange interaction of the antiferromagnetic state. The electrical resistivity at zero field shows that these compounds are electrically insulating materials, which can be explained by the predominance of the antiferromagnetic ordering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The Ruddlesden–Popper (RP) phases with general formula T2–2xD1 + 2xM2O7 (where T is the trivalent rare earth cation such as La, Sm, Pr, etc., D is the divalent alkaline earth cation such as Sr, Ca, Ba, etc., and M is a transition metal such as Mn, Fe, Co, etc.) [1,2,3,4,5,6,7,8,9,10] have attracted considerable research interest in recent years due to their interesting electrical, magnetic, and electrochemical properties [11,12,13,14,15,16], including magnetic storage and spintronics [17, 18]. Moreover, they have garnered increasing attention as oxygen catalysts due to their compositional flexibility, distinctive layered structure, and exceptional intrinsic activity [19]. Furthermore, these materials have been investigated as solid electrolytes for lithium-ion batteries to tackle safety and performance issues associated with conventional liquid electrolytes. They offer advantages such as high ionic conductivity, stability, and compatibility with lithium metal anodes [20]. The general formula of the RP phases could also be written as (T, D)n + 1MnnO3n + 1 consists of RP phases T1−xDxMnO3 blocks which are n octahedra thick and separated by rock-salt DO layers. The simple perovskite structure is with n = ∞ in the RP series. Several studies have been carried out about RP manganates for n = 2 such as the (La2–2xCa1 + 2xMn2O7) series [12]. It has been shown that the magnetic and electrical properties of the RP compound with n = 2 are very sensitive to the chemical nature of T and D ions [13]. Systematic studies on T2–2xD1 + 2xMn2O7 materials have been devoted to examining the structural, electrical, and magnetic mechanisms that control the reported properties. Mixed valence Mn3+/Mn4+ is very important to understand these mechanisms. We noted that, first, the origin of these transitions is attributed to the double-exchange or super-exchange interactions between the manganese cations and oxygen, which are generally explained by the ferromagnetic and antiferromagnetic coupling between Mn3+(t2g3eg1) and Mn4+(t2g3eg0) cations and second, the structure defect such as deformation of the MnO6 octahedra, which implies a distortion of the structural network due to the Jahn–Teller effect [21,22,23,24]. Several studies on the RP phases based on samarium of the formula Sm2–2xA1 + 2xMn2O7 (A: alkaline earth) [25,26,27,28] have reported the existence of an antiferromagnetic transition below Néel temperature (TN). Moreover, other similar compounds exhibit a double magnetic transition at low temperatures such as the Sr1.6Sm1.4Mn2O7 compound [28]. To our knowledge, no compound of the RP phases (n = 2) based on samarium with a mixture of two alkaline earth metals (barium and strontium) has been reported. In this work, we report a synthesis approach of manganates Sm1.5–xSr1 + xBa0.5Mn2O7 (with x = 0, 0.1, and 0.3). Their crystal structures were characterized by X-ray diffraction (XRD) using the Rietveld method refinement. The electrical and magnetic properties of bilayered manganates are also investigated.
2 Experimental
2.1 Synthesis protocol
Poly-crystalline compounds Sm1.5–xSr1 + xBa0.5Mn2O7 with x = 0, 0.1, and 0.3 were synthesized by the co-precipitation method from stoichiometric amounts of samarium oxide Sm2O3 (99.99% Purity), strontium nitrate Sr(NO3)2 (99.99% Purity), barium nitrates, Ba(NO3)2 (99.99% Purity) and manganese nitrate tetrahydrate Mn(NO3)2.4H2O (99.99% Purity). The reagents are dissolved simultaneously in 100 mL of distilled water, 10% nitric acid is added to promote their solubilization, then the mixtures are stirred at 343 K for 24 h, and the obtained precipitates are introduced into a platinum crucible for heat treatment. The conventional sintering was carried out under air in a muffle furnace. The first annealing step was carried out at 973 K for 24 h to decompose the residual nitrates. The obtained powders are ground in an agate mortar then pelletized using a hydraulic press, and brought to temperatures of 1173 K, 1473 K, and 1673 K, respectively, for the second, third, and fourth heat treatments. Each heat treatment lasts 24 h and is interspersed with an intermediate step of grinding and pelletizing.
2.2 Instrumental analyses
The analysis of the products was carried out using X-ray powder diffraction. The X-ray data were collected at room temperature on a BRUKER D8 Advance (θ-θ) diffractometer with CuKα radiation (40 kV, 40 mA). XRD patterns were recorded in the range of 2θ = 10°–100° with a 0.01° step. Rietveld refinement was performed using the Fullprof program. The crystallite size and micro-strain were estimated using Williamson–Hall (WH) plots for FWHM of XRD patterns after Rietveld fitting. The magnetic measurements were conducted in the temperature range of 2–275 K at a magnetic field of 0.05 T, using a BS2 magnetometer developed at Neel Institute. The magnetic measurements versus the magnetic field varying from 0 to 5 T were also measured. The electric resistivity of samples was measured using a source measure Keithley 236 with a standard four-probe technique with an Ag electrode in the temperature range of 150–320 K.
3 Results and discussion
3.1 Structural characterization and refinement
The structure of Sm1.5–xSr1 + xBa0.5Mn2O7 (x = 0, 0.1, and 0.3) samples has been refined on powder X-ray diffraction patterns by the Rietveld refinement method using Fullprof software. The pseudo-Voigt function was used to describe the lines’ shape of the diffraction patterns. The shift errors parameter, unit cell parameters, background coefficients, the Caglioti coefficients U, V, and W, atomic positions, isotropic shift parameters, and preferred orientation were successfully refined. The occupancy rate parameters are fixed according to the ideal stoichiometry of the crystallographic site. The experimental and calculated X-ray diffraction patterns after full refinement are shown in Fig. 1 (a). Indeed, all these parameters, including the unit cell parameters, reliability factors, atomic positions, and thermal displacement parameters, are presented in Table 1. The XRD diagrams were fitted using a pseudo-Voigt function and a third-order polynomial was employed to assimilate the background function. The results show a good agreement between the observed and the calculated diffraction profiles, with good reliability refinement factors (Table 1). The results show that all samples present single phases and all diffraction peaks for each phase are indexed in the tetragonal system with the limiting condition for reflection h + k + l = 2n and I4/mmm as space group [29, 30]. Two distinguished crystallographic sites exist in the structure of these compounds that can host Sm3+, Ba2+, and Sr2+ cations. The first one is the perovskite cage of dodecahedral coordination with Wyckoff site 2b, and the second one is a nine-coordinated with Wyckoff site 4e. The manganese ions Mn3+ and Mn4+ are located in an octahedral environment with Wyckoff site 4e. Figure 1b shows the structure schema of Sm1.5–xSr1 + xBa0.5Mn2O7 generated by ATOMS software.

a Rietveld refinement patterns of Sm1.5–xSr1 + xBa0.5Mn2O7. The upper symbols illustrate the observed data (circles) and the calculated pattern (solid line). The vertical markers show the calculated positions of Bragg reflections. The lower curve is the difference diagram. b Crystal structure schema of Sm1.5–xSr1 + xBa0.5Mn2O7
Several studies have proven that the substitution of an alkaline earth ion by a rare earth ion occurs preferentially at the DO layered with (D = alkaline earth ion or rare earth) [31,32,33]. However, the small size of the rare earth ions compared to the alkaline earth ions leads to a privileged occupation of the latter in the perovskite cage. The lengths of the bonds and the angles between the atoms in the studied structures adopt values conventionally encountered in previous studies [30,31,32,33]. Bond lengths and angles for compounds x = 0, 0.1, and 0.3 are listed in Table 2. The lengths of the equatorial Mn–O(2) bonds are shorter than the apical Mn–O(1) and Mn–O(3) bonds and the lengths of the apical Mn–O(1) bonds are shorter than those of the Mn–O(3)bonds, this is due to the existence of a distortion at the MnO6 octahedra. Therefore, the Mn ions have five neighboring oxygen anions (O(1) and 4O(2)), precisely four oxygen atoms (4O(2)) distant at the same distance, and the oxygen atom O(3) is further away, which implies that the MnO6 octahedra are almost irregular. The origin of this deformation may be due to the Jahn–Teller effect generated by the Mn3+ (t2g3eg1) ions.
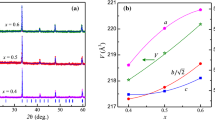
Figure 2a. shows the evolution of a and c lattice parameters versus the strontium substitution rate and Fig. 2b. shows the volume unit cell versus the strontium substitution rate. To fully explain the evolution of unit cell parameters, two main factors are taken into account: the first one is the cations ionic radii of Sm+3 and Sr2+, and the second one is the oxidation of Mn3+ in to Mn4+ due to the substitution of samarium by strontium according to the following coupled substitution: Sm3+ + Mn3+ → Sr2+ + Mn4+.The evolution of a unit cell is governed by the ionic radii of both Sr2+ and Sm3+cations. At equal coordinates, the cationic radius of strontium is higher than that of samarium [34]; consequently, a weak expansion occurs in the ab plane; therefore, a unit cell parameter increases weakly with the strontium substitution rate.

a Evolution of the unit cell parameters vs. substation rate, b evolution of volume of the elementary cell vs. substation rate, c octahedron distortion vs. substation rate, and d MnO6distorted octahedra schema
The evolution of the c parameter and unit cell volume as a function of strontium amount, are strongly correlated. Owing to the preservation of local electro-neutrality, the substitution of Sm3+ by Sr2+ generates partial oxidation of manganese Mn3+ to Mn4+. Further, the decrease of Mn3+ ions amount normally implies a decrease in Jahn–Teller type distortion; this leads to the compression of the apical Mn–O bond along the “c” axis. Consequently, the c parameter decreases with the strontium substitution rate. The MnO6 octahedral distortion (Δ) [28] has been calculated by the following Eq. (1) and the results are shown in Fig. 2©.
with d(Mn–Oequatorial) as the equatorial bond distance in MnO6 octahedra and d(Mn–Oapical) as the average distance of two apical bonds (Fig. 2(d). The compound with x = 0 having a 75% of Mn3+, with a high strain (Δ = 1.065), and for x = 0.1 having 70% in Mn3+ exhibits a slightly lower value of strain than that of the first compound (Δ = 1.056), finally for x = 0.3 (60% in Mn3+) is characterized by an even lower strain (Δ = 1.045) than that of compounds with x = 0 and x = 0.1. The substitution of Sm3+ by Sr2+ generates partial oxidation of manganese Mn3+ to Mn4+ and, thus, a decrease in the Jahn–Teller effect type distortion, which leads to the reduction of MnO6 octahedral deformation. The crystallite size and lattice strain were calculated using a WH plot [35, 36] following Eq. (2):
where D is the crystallite size (nm), k is a Scherrer’s constant (0.94 for spherical particles), λ (nm) is the wavelength, β (rad) is the full width at half of the maximum (FWHM) of the intense and broad peaks, θ is the Bragg angle and ε is the average value of lattice strain. Figure 3 illustrates the WH plot of Sm1.5–xSr1 + xBa0.5Mn2O7, (a) for x = 0, (b) for x = 0.1, and (c) for x = 0.3. The values of mean crystallite size are about (45 ± 5) nm; it is noted that the size of the crystallites does not change with the substitution rate. The correspondence to the obtained microstrain by WH plot for these samples is very small (ε = 0.11%). Furthermore, the WH plot showed that the compounds do not suffer from a pronounced microstrain.

The Williamson–Hall plot of Sm1.5–xSr1 + xBa0.5Mn2O7, a for x = 0, b for x = 0.1, c for x = 0.3
3.2 Magnetic properties
The magnetic measurements versus temperature were carried out for the polycrystalline samples Sm1.5–xSr1 + xBa0.5Mn2O7 x = 0, 0.1, and 0.3. The evolution of magnetization measurement versus temperature at magnetic field 0.05 T is shown in Fig. 4a–c, respectively, for x = 0, 0.1, and 0.3 compounds.

Magnetization vs. temperature at magnetic field 0.05 T (a), (b), and (c), respectively, for the compounds x = 0, x = 0.1, and x = 0.3, in system Sm1.5–xSr1 + xBa0.5Mn2O7. d variation of the magnetization vs. field at 5 K
One can observe that magnetization indicates the existence of the predominant antiferromagnetic order at low temperatures for all compounds. Therefore, these compounds' magnetic behavior is fundamentally governed by a single magnetic transition established at the Neel temperature (TN) of approximately 25 K. The antiferromagnetic behavior present in these materials can be essentially explained by the super-exchange interactions through the π bond of the O2− ion between the t2g orbitals of neighboring ions of Mn3+and Mn4+ (t2gMn4+–π(O2−)–t2gMn3+) lead to an antiparallel alignment of magnetic moments. Moreover, surface defects and impurities can introduce local disturbances in the magnetic order and influence global magnetic behavior. Additionally, hybridization can also affect the number and distribution of electrons involved in the exchange interaction as a result it can modify the strength of the exchange interaction or lead to the formation of different magnetic phases and finally affect magnetic response. The magnetic behavior of these compounds presents a true analogy with most of the phases rich in samarium [26, 27]. The isothermal magnetization measurements versus field MH from 0 to 10 T were carried out for the compounds x = 0, x = 0.1; and x = 0.3 at 5 K. The results are shown in Fig. 4d. It can be seen that at 5 K the isotherms for the three compounds studied show similar MH patterns behavior. The MH curves of the three compounds show a slight curvature at weak fields, showing the saturation is never reached even with a strong magnetic field ≈ 10 T. This phenomenon confirms the presence of strong super-exchange interactions of the antiferromagnetic state at low temperature [37]. The absence of saturation at 5 K despite a high magnetic field ≈of 10 T indicates a strong antiferromagnetic order at low temperatures. Similar behavior has been observed in CaMn2O4 [38] and Er2Mn2O7 [39] compounds.
3.3 Electric properties
The electrical resistivity measurements of both compounds x = 0.1 and x = 0.3 were made using the four-point method. Figure 5a represents the thermal evolution of their electrical resistivity under zero field. We notice that, below 315 K, the resistivity increases while the temperature decreases, indicating a progressive localization of charge carriers in the form of small magnetic polarons [40,41,42,43]. This confirms the existence of an electric insulating behavior in these compounds.

a The thermal evolution of the electrical resistivity vs. temperature under zero field and b curves Lnρ vs. f (1/T) for Sm1.5–xSr1 + xBa0.5Mn2O7 compounds for x = 0.1 and x = 0.3
The electrical behavior characterizing these compounds is similar to that presented in previous studies for the polycrystalline phases associated with antiferromagnetic interactions.[28, 44]. The electrical behavior in the high-temperature insulating regime is usually represented by the first approximation considering that the resistivity is governed by a classical thermal activation law, in which the resistivity is expressed by the following Eq. (3):
where Ea is the activation energy of the small charge carrier, ρ0 is a constant (the electrical resistivity at an infinite temperature), kB is the Boltzmann constant, and T is the temperature. The activation energy was calculated following a linear form of the activation law expressed by the following Eq. (4):
The curves Lnρ vs. 1/T for the compounds with x = 0.1 and x = 0.3 are shown in Fig. 5b The values of the activation energy of the compounds with x = 0.1 and x = 0.3 are, respectively, 133 and 135 meV. The activation energy is found to increase slightly with the increase of the strontium rate. This phenomenon can be explained by the decrease in charge carrier activation, which usually decreases with the incorporation of strontium into the crystal structure, thus stabilizing the insulating state of these oxides. This behavior is also confirmed by Liu et al. [27]. In addition, this behavior can be explained considering the structural aspect by a reduction of the average distortion of the octahedral network MnO6 due to the incorporation of Mn4+ ions within the network. Therefore, the decrease in the intensity of the cooperative Jahn–Teller effect favors the degeneration of the orbital eg and t2g, which generates an increase in the activation energy of the charge carriers. It is noted that the Mn3+/Mn4+ ratio decreases by increasing the strontium amount, which leads to a decrease of eg orbital electrons, weakening the double-exchange mechanism.
4 Conclusion
The co-precipitation route successfully synthesized the manganates Sm1.5−xSr1+ xBa0.5Mn2O7 compounds with x = 0, 0.1, and 0.3. The structural refinement results show that all samples present single phases and all diffraction peaks for each phase are indexed in the tetragonal system iso-structural phase of Sr3Ti2O7-type structure with I4/mmm as a space group. Magnetic measurements vs. temperature indicate the existence of the predominant antiferromagnetic order of super-exchange interactions at low temperatures for all compounds. The magnetic behavior of prepared materials is governed by a single magnetic transition established at the TN ≈ 25 K. The electrical resistivity measurements for x = 0.1 and x = 0.3 show that both compounds have an insulating behavior which is due to the predominance of the antiferromagnetic ordering.
Data availability
All data included in this study are available upon request by contact with the corresponding author.
References
R. Zhang, A.S. Gibbs, W. Zhang, P.S. Halasyamani, M.A. Hayward, Inorg. Chem. 56, 9988 (2017)
K. Wissel, T. Vogel, S. Dasgupta, A.D. Fortes, P.R. Slater, O. Clemens, Inorg. Chem. 59, 1153 (2019)
H.A. Hopper, E.J. Wildman, D.E. Macphee, F. Sher, C. Ritter, M. Lledos, G. Stenning, A.C. McLaughlin, J. Solid State Chem. 253, 313 (2017)
W. Zhong, X.S. Liu, H.Y. Jiang, L.J. Zhao, Y.W. Du, N. Zhang, Appl. Phys. A 78, 575 (2004)
S.E. Ouahbi, M. Lassri, M. Sajieddine, H. Lassri, Appl. Phys. A 128, 632 (2022)
Y. Ounza, S.E. Ouahbi, R. Moubah, M. Lassri, M. Sajieddine, M. Oubla, I. Abdelrhafor, E.K. Hlil, H. Lassri, Appl. Phys. A 129, 418 (2023)
M. Oubla, M. Lamire, H. Lassri, E.K. Hlil, MATEC Web of Conferences (EDP Sciences, 2013), p.04040
Y. Ounza, M. Bouhbou, M. Oubla, M. Moutataouia, M. Lamire, E.K. Hlil, H. Lassri, J. Supercond. Novel Magn. 33, 3791 (2020)
Y. Ounza, R. Moubah, M. Oubla, A. Elouafi, M. Moutatouia, M. Lamire, E.K. Hlil, H. Lassri, Solid State Commun. 350, 114785 (2022)
A. Elouafi, Y. Ounza, L.H. Omari, M. Oubla, M. Lassri, M. Sajieddine, H. Lassri, Appl. Phys. A 127, 1 (2021)
A. Ramirez, J.R. Cavanaugh, J.M. Purcell, Cond. Mat 9, 8171 (1997)
H. Asano, J. Hayakawa, M. Matsui, Appl. Phys. Lett. 68, 3638 (1996)
M.-H. Phan, S.-C. Yu, J. Magn. Magn. Mater. 308, 325 (2007)
S. Vasala, A. Jakob, K. Wissel, A.I. Waidha, L. Alff, O. Clemens, Adv. Electron. Mater. 6, 1900974 (2020)
Y. Zhang, J. Wang, P. Ghosez, Phys. Rev. Lett. 125, 157601 (2020)
B.M. Silva, J. Oliveira, T. Rebelo, V.B. Isfahani, P. Rocha-Rodrigues, N. Lekshmi, J.H. Belo, F.L. Deepak, A.M.L. Lopes, J.P. Araújo, Mater. Res. Bull. 158, 112066 (2023)
A. El Hachmi, F. El Bachraoui, S. Louihi, Y. Tamraoui, S. Benmokhtar, L. Bih, M. Sajieddine, P. Lazor, B. Manoun, J. Inorg. Organomet. Polym Mater. 30, 1990 (2020)
A. Pal, A. Rao, D. Kekuda, B.S. Nagaraja, R. Mondal, D. Biswas, J. Magn. Magn. Mater. 512, 167011 (2020)
G. Pan, Y. Deng, L. Zhao, H. Wang, R. Wang, J. Jin, Y. Gong, B. He, Electrochim. Acta 424, 140673 (2022)
X. Liu, L. Gu, Small Methods 2, 1800006 (2018)
P.M. Chowdhury, A.K. Raychaudhuri, Mater. Res. Bull. 137, 111160 (2021)
A. Karati, T. Parida, J. Gupta, H.K. Adigilli, P.H. Borse, J. Joardar, Mater. Res. Bull. 137, 111181 (2021)
G.M. Kuz’micheva, L.I. Ivleva, I.A. Kaurova, E.V. Khramov, V.B. Rybakov, M.E. Doroshenko, Mater. Res. Bull. 140, 111300 (2021)
H.-C. Chen, D.-J. Jan, B.-C. Lin, T.-H. Hsueh, Mater. Res. Bull. 140, 111313 (2021)
M. Oubla, M. Lamire, H. Lassri, S. Benmokhtar, E.K. Hlil, J. Supercond. Novel Magn. 27, 1715 (2014)
J.W. Liu, G. Chen, Z.H. Li, Z. Lü, Z.G. Zhang, Mater. Chem. Phys. 105, 185 (2007)
J.W. Liu, G. Chen, Z.H. Li, Z.G. Zhang, Solid State Commun. 141, 341 (2007)
S. Biswas, S. Chatterjee, P. Chatterjee, P. Dutta, A.G. Joshi, A.K. Nigam, S.K. De, S. Chatterjee, Phys. Rev. B 66, 214403 (2002)
S.N. Ruddlesden, P. Popper, Acta Crystallogr. A 11, 54 (1958)
M. Oubla, M. Lamire, A. Boutahar, H. Lassri, B. Manoun, E.K. Hlil, J. Magn. Magn. Mater. 403, 114 (2016)
D. Singh, A. Mahajan, Ceram. Int. 41, 15048 (2015)
P.D. Battle, M.A. Green, N.S. Laskey, J.E. Millburn, L. Murphy, M.J. Rosseinsky, S.P. Sullivan, J.F. Vente, Chem. Mater. 9, 552 (1997)
A. Belkahla, K. Cherif, J. Dhahri, E.K. Hlil, J. Supercond. Novel Magn. 29, 19 (2016)
R.D. Shannon, Acta Cryst. A 32, 751 (1976)
A. Baidya, A. Dutta, Mater. Res. Bull. 160, 112114 (2023)
U. Nissar, J. Ahmad, S.H. Bukhari, Y. Iqbal, Mater. Res. Bull. 127, 110844 (2020)
J.W. Liu, G. Chen, Z.H. Li, W.W. An, Z.G. Zhang, J. Alloy. Compd. 431, 1 (2007)
S. Zouari, L. Ranno, A. Cheikh-Rouhou, O. Isnard, M. Pernet, P. Wolfers, P. Strobel, J. Alloy. Compd. 353, 5 (2003)
N.B. Amor, M. Bejar, E. Dhahri, M.A. Valente, J.L. Garden, E.K. Hlil, J. Alloy. Compd. 563, 28 (2013)
D.N. Argyriou, H.N. Bordallo, B.J. Campbell, A.K. Cheetham, D.E. Cox, J.S. Gardner, K. Hanif, A. Dos Santos, G.F. Strouse, Phys. Rev. B 61, 15269 (2000)
N.I. Solin, J. Magn. Magn. Mater. 401, 677 (2016)
D. Singh, M. Sharma, S. Sharma, R. Singh, Bull. Mater. Sci. 34, 1089 (2011)
H. Chang, Q. Wu, Appl. Phys. A 115, 1323 (2014)
S. Valencia, L. Peña, Z. Konstantinovic, L. Balcells, R. Galceran, D. Schmitz, F. Sandiumenge, M. Casanove, B. Martínez, J. Phys. Condens. Matter 26, 166001 (2014)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Oubla, M., Ounza, Y., Lamire, M. et al. Investigation of structural, magnetic, and electrical properties of layered perovskite Sm1.5–xSr1 + xBa0.5Mn2O7 (with 0 ≤ x ≤ 0.3). Appl. Phys. A 129, 574 (2023). https://doi.org/10.1007/s00339-023-06852-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-023-06852-2