
Abstract
Herein, we present our numerical results of ZrxSi1-xO2 solid solutions (with x = 0–1) in order to predict new tetragonal hypothetical compounds from Zircon family. We have used the full-potential linearized augmented plane waves method based on the density functional theory DFT within GGA approximation as implemented in WIEN2k code to investigate the structural and optoelectronic properties of the named compounds. We have studied, in first step, the properties of the ZrSiO4 compound, where the obtained results are compared to several theoretical and experimental investigations. Secondly, we have predicted new hypothetical compounds for the Zircon family ZrSi3O8 and Zr3SiO8 which crystallize in I41/amd space group. Whatever, the chemical stability of the alloys under study was checked with both formation enthalpy and cohesive energy. Furthermore, the phonon dispersion results indicated this aspect also. The electronic properties reveals a strong p-d covalent hybridization between Zr and O. The Zr3SiO8 compound exhibits a large indirect band gap, while the ZrSi3O8 presents a direct band gap of 2.008 eV at Γ point. The optical quantities inspired from the dielectric function calculation are also calculated and discussed in details.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The zircon family alloys are indispensable materials for their diverse technological applications not only in electronics field but also in metallurgical industry and chemical industry [1, 2]. The zirconium oxide compound (ZrO2) is used to manufacture a catalyst system for the desired carbon dioxide conversion and others chemicals reactions [2]. In the recent years, the zirconium silicate alloys have been attracted the attentions of the experimental and the theoretical researcher groups [3,4,5] due to their interest electronic and optical properties. One of the most zircon family alloys studied is the ZrSiO4 which it can be crystallized on two phases according the pressure value, the first one is the zircon phase at ambient pressure (at low pressure) but when compressed under hydrostatic pressure it converts into scheelite phase. The scheelite type ZrSiO4 was first synthesized by Reid and Ringwood [6] and was confirmed by Liu [7] at pressure up to 20 GPa. The ZrSiO4 has a wide band gap (in order of 4.43 eV), large band offsets, low thermal conductivity and low coefficient of thermal expansion [8] these useful proprieties make it a good candidate for high-temperature devices and anti-oxidation protective coatings [9,10,11]. On the other hand, the measurement of the optical properties shows that the ZrSiO4 has a high dielectric permittivity (in order 7.9–13), a high refractive index (in order 1.93–2.01) and exhibits a strong dispersion which makes it as preferred opacifier for the ceramic glaze industry [12], those important characteristics can be used to manufacturing electronic devices to avoid the direct tunneling current induced by the continuous minimization of complementary metal-oxide semiconductor devices [13]. The most apparent interest on this compound is the presence of Si atom that makes them interfaced with silicon system with high stability [14].
Other stoichiometric materials derivative of the zirconia family alloys, the ZrxSi1-xO2 alloys, where x is the variable-composition between Zr and Si atom, at the limit (X= 0 and 1) all possible is ZrO2–SiO2 compounds. Among a number of crystalline forms of ZrO2–SiO2 at ambient pressure and zero Kelvin, Zhang et al. [1] reported that the ZrSiO4, Zr3SiO8 and ZrSi2O6 are stable or metastable crystal structures of zirconium silicates. The ZrSi2O6 and Zr3SiO8which can also be an axis of research, some papers in the literature show that these compounds have a significantly ionic bonding character, a high refractive index (~ 2.4) and a higher dielectric constant compared to the other Zr-based materials and are insulators [1, 2]. If the dielectric constant, band gap and breakdown field are simultaneously optimized these compounds will be an excellent energy storage material for electronic applications and give a new feature to investigation a high dielectric material.
Today and by the mean of the density functional theory (DFT), we can investigate the physical properties of these varieties of materials and gives good estimation results to confirm the useful one in experiment works. The research of the electronic properties of derivatives zirconium silicates presents a wide field of investment not only for the physics of materials but also for general physics. It can be used also as a solid electrolyte in oxygen scavengers [2], as thermal barrier coating applications [8] and as nuclear waste containment [15].
In the literature, the physical and chemical properties of zircon (ZrSiO4) have been investigated in great detail with theoretically and experiment study but a few works have been reported about ZrxSi1-xO2. In order to exploit it in the technological domain, a best knowledge of its properties is necessary, that motived us to investigate, in great detail, the physical proprieties of these alloys.
In this paper, we present our study on the structural and the optoelectronic proprieties of ZrxSi1-xO2 alloys for x = 0.25, 0.50 and 0.75. In first step, the crystalline structure of the compound ZrSiO4 is considered as reference and by varying the rate x, we predict the new tetragonal hypothetical ZrSi3O8 and Zr3SiO8 compounds, where we focus our attention also to find the most stable structure through the calculation of the energy total of the ground state, thermodynamic stability and dynamic stability. Secondly, we investigate their optoelectronic properties in the objective to know their potential applications.
2 Calculation method
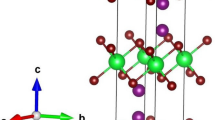
For the present study, we employed the WIEN2k package [16] in the framework of the density functional theory (DFT) and based on the full potential-linearized/augmented plane wave (FP-L/APW) method to investigate the optoelectronic properties of our compounds. The generalized gradient approximation (GGA) established by Perdew-Burke-Ernzerhof (PBE) [17] was used for the exchange correlation potential. Some relevant input parameters for the self-consistent field (SCF) initialization are taken from the convergence test such as muffin-tin (MT) spheres radius and number of k-point in the irreducible Brillouin zone (IBZ), etc. For the basis function the \({R}_{\mathrm{MT}}{K}_{\mathrm{max}}\) is taken equal to 7 (where \({K}_{\mathrm{max}}\) is the maximum modulus for the reciprocal lattice vector and \({R}_{\mathrm{MT}}\) is the smallest MT sphere radius). For non-overlapping MT spheres, the RMT values are 1.91, 1.28 and 1.49 Bohr for Zr, Si and O atoms, respectively. The maximum angular momentum for the wave function expansion inside the muffin-tin sphere was confined to lmax = 10. The charge density was Fourier expended up to Gmax = 12. The Monkorst-Pack special k-points in the (IBZ) is performed for 102 k-points of 10 \(\times\)10 \(\times\)10 meshes which equivalent to 1000 k-points in BZ. The cut off energy, which defines the separation of valence and core states, was taken as − 6.0 Ry for all compounds and the self-consistent calculations are considered to be converged when the total energy of the crystal is converged to10−4 Ry. The unit cell for these compounds are taken in tetragonal crystalline structure with the space group I41/amd (N = 141), (Fig. 1).

Crystal structure of a ZrSi3O8, b ZrSiO4 and c Zr3SiO8
3 Results and discussions
3.1 Structural properties and thermodynamic stability
In order to predict a new compound from zircon family, we have studied the ZrxSi1-xO2 solid solutions for the rate x = 0.25, 0.5 and 0.75 which corresponding the ZrSi3O8, ZrSiO4 and Zr3SiO8 alloys. The structural properties are investigated by the calculation of the total energy as a function of the unit cell volume around the equilibrium volume V0. Figure 2 shows the volume optimization curves obtained for each compound, whereas the data are fitted by the following Murnaghan's equation of state [18] in order to calculate the equilibrium lattice parameters a, c and the bulk modulus and its derivative \({B\mathrm{ and }B}_{p}\), respectively:
where, \(E\left(V\right)\) is the total energy,\(\mathrm{V}\) is the unit cell volume.

Volume optimization for ZrSi3O8, ZrSiO4 and Zr3SiO8 compounds using GGA approximation
The obtained results are summarized in Table 1 together with the theoretical and experimental data reported in the literature. In view of this table, the lattice parameters (a and c) are directly affected by the variation of the zirconium concentration (x), we can see that the lattice parameters increased when the zirconium concentration increases, this variation is due to the difference between the atomic radius of zirconium (RZr = 1.6 Å) compared to that of substituted atom of silicon (RSi = 1.1 Å). The increase in the size of the compounds is accompanied by the decrease in the bulk modulus when the rate of zirconium is enhanced. The bulk modulus informs us about the rigidity of the crystal and its resistivity to the mechanical strain. In fact, for greater value of bulk modulus, the materials loss their rigidity and become less resistant to the shear strain. According to our results (See Table 1), the ZrxSi1-xO2 materials become rigid and more compressible when the zirconium concentration increases. Generally, the calculated ground state properties of ZrSiO4 alloy (x = 0.5), such as the equilibrium lattice constants a, c and the bulk modulus are in good agreement with the available experimental and theoretical results in the literature. Where, in the best of our knowledge, there are no experimental or theoretical data available for the other alloys (ZrSi3O8 and Zr3SiO8), hence our results are the first theoretical prediction for these materials.
To ensure the stability of the titled compounds, we have examined the thermodynamic stability through the calculation of the cohesive energy (Ecoh) and the formation enthalpy (ΔHfor) using the following formula [23, 24]:
where: \(E_{{{\text{tot}}}}^{{{\text{Zr}}_{x} {\text{Si}}_{1 - x} {\text{O}}_{2} }}\) is the total energy for the titled alloys. The (\(E_{{{\text{Zr}}}}^{{{\text{atom}}}}\), \(E_{{{\text{Si}}}}^{{{\text{atom}}}}\) and \(E_{{\text{O}}}^{{{\text{atom}}}}\)) are the total energy of (zirconium, silicon and oxygen) atoms, (\(E_{{{\text{Zr}}}}^{{{\text{solid}}}}\), \(E_{{{\text{Si}}}}^{{{\text{solid}}}}\) and \(E_{{{\text{O}}_{2} }}^{{{\text{gas}}}}\)) also the total energy of (solid zirconium, solid silicon and oxygen gas), respectively, in their whole structures.
All the mentioned quantities in formula are obtained via performing an accurate potential of GGA-PBE-sol-06 approximation [25]. The obtained values of both Ecoh and ΔHfor are summarized in (Table 1). It seems clear that all the predictive results with negative signs which indicate that the titled alloys in good thermodynamic stability, moreover, these alloys could be synthesize easily at laboratory [23, 24].
To understand the dynamical stability of herein studied alloys above knowing their thermal behavior, we have investigated the phonon propriety using the Phonopy package [26] interfaced with the WIEN2k code. An atom of the crystal is displaced from its equilibrium position and obtaining perturbed crystal structures, and the lattice dynamical properties were analyzed in term of phonon frequencies as a second-order derivative of the total energy with respect to atomic displacements [27, 28]. In the phonon dispersions curves, there are two modes of phonons, the optical mode and the sonic mode. The optical mode (a higher frequency) is obtained when two different adjacent atoms move against to each other, while in the sonic mode (a lower frequency), they move together.
The phonons are quantum lattice vibrations formed by the collective motion of many atoms in a crystal lattice. The total energy of the phonons is calculated using the following equation:
\(w\) is the angular frequency of the vibration, \(n\) is the number of phonons and E is the total energy. In order to predict the stable and metastable phase of these compounds, we have used to calculate the phonon dispersion curves using a supercell with 24 atoms and 200 k-point. Figure 3 shows that our calculations confirm that zircon is the only thermodynamically stable zirconium silicate and the absence of imaginary phonon frequencies part verifies with success that the phase (I41/amd-ZrSiO4, I41/amd-Zr3SiO8 and I41/amd-ZrSi3O8) are dynamically stable.

Phonon dispersion curves for ZrSi3O8, ZrSiO4 and Zr3SiO8 at ambient pressure
3.2 Electronic properties
In the following section, we will present our obtained result on the electronic properties which are mainly determined by the calculation of the band structures, the density of states and the charge densities, this quantities allow us to deduce the nature and the value of the gap energy, the nature of the chemical bonds between the different elements constituting the material, the orbital contribution of each atom in the conduction and valence bands and what states are dominate in the both bands.
3.2.1 Band structure and density of states
We present the electronic structure of the studied compounds for a crystalline structure with space groupI41/amd. The electronic bands structures are calculated along the high symmetry points of the first Brillouin zone (points \({\Gamma ,}\;{\text{X,}}\;{\text{M}}\) and \({\text{R}}\)) as well as the total and partial densities of states are projected on the same energy axis. In Figs. 4, 5 and 6, we displayed together the bands structures and the total/partial densities of states of ZrSi3O8, ZrSiO4 and Zr3SiO8 alloys, respectively.

Band structure and total / partial densities of state (TDOS / PDOS) of ZrSi3O8 compound using GGA approximation

Band structure and total / partial densities of state (TDOS / PDOS) of ZrSiO4 compound using GGA approximation

Band structure and total / partial densities of state (TDOS / PDOS) of Zr3SiO8 compound using GGA approximation
From curves of the partial densities of states (PDOS), we can observe that the valence band of Zr3SiO8, ZrSi3O8 and ZrSiO4 is mainly dominated by the p-O (oxygen) state with a weak contribution of p-Si and d-Zr states, where we can see clearly from partial DOS of all our alloys that the conduction band is constructed by the high contribution of d-Zr state with p-O orbital and a weak contribution of p-Si state. In detailed view of PDOS,TDOS and the band structure, at the bottom of the Fermi level, for Zr3SiO8 alloys the main peak (A) of TDOS is located at − 0.75 eV which is induced by the presence of the 2p orbital of oxygen atom, the peak (B) situated at − 2.71 eV is constructed by the mixture of the d orbital of Zr atom and p orbital of oxygen atom, and the peak (C) situated at − 5.98 eV is formed by the presence of p orbital of oxygen and silicon atom. Above Fermi level, the main peak (D) in the band conduction is located at 4.22 eV, this peak is mainly formed by the contribution of d-Zr and p-O orbital. In addition, we can deduce that the p-O orbital and d-Zr are responsible for the electronic transition from valance to conduction band with an indirect large band gap (Γ-M) of 3.675 eV.
For ZrSi3O8 alloy there are two important peaks, the first one in valance band is the peak (A) which is located at − 0.75 eV and constructed by the mixture of d-Zr and p-O orbital, the second one in conduction band is the peak (B) which is at 3.34 eV and formed by the hybridization of d-Zr, p-Si and p-O orbital. For this alloy (ZrSi3O8), the p-O, d-Zr and p-Si orbital are responsible for the electronic transition from occupied state to unoccupied state, with direct band gap (Γ-Γ) of 2.008 eV.
For ZrSiO4 alloy, we see clearly that its valance band is dominated principally by d-Zr and p-O orbital, where this later create a peak (A) at − 0.90 eV. While its conduction band has a peak (B) located at 4.72 eV, which is formed by same orbital of valance band (d-Zr and p-O). ZrSiO4 have an indirect band gap of 4.565 eV sited at Γ-M direction.
To understand further the effect of the zirconium rate (x) on the band gap of ZrxSi1-xO2 alloys, we have plotted, in Fig. 7, the band gap versus the rate x for x = 0.25, 0.5 and 0.75, we can see clearly that the band gap of ZrSiO4 (corresponding x = 0.50) is reduced when the rate of zirconium is changed to 0.25 or 0.75, this result shows that the band gap of ZrxSi1-xO2 alloys can be optimized to the desired value by the variation the zirconium concentration. The band gap value of herein studied alloys is summarized in Table 2 together with the theoretical and experimental data available in the literature. It seems that our obtained results are in good agreement with those investigations.

Gap energy versus x concentrations
3.2.2 Charge densities
In order to examine the bond character, the charged vacancies and the charge transfer in ZrSiO4, Zr3SiO8 and ZrSi3O8 alloys, we have investigated the projection of the charge densities on hkl plans, using GGA approximation. The obtained results are displayed in Fig. 8. The results of herein studied alloys are topologically similar. These results show that the distribution of charge density is spherical around the Zr atom which illustrates that the d-Zr level is entirely filled, where the distribution of charge densities around Si atom are roughly spherical, which illustrates that the p-Si levels are partially filled. Generally, there are over lapping of the electron density between the states of Zr, Si and O atoms which indicating the covalent nature of bonds between these atoms. The covalent bond can be explained by the high rigidity of our compounds.

Charge densities of ZrSi3O8, ZrSiO4 and Zr3SiO8 compounds using GGA approximation
3.3 Optical properties
The optical properties of solids are a very important for the fundamental research and the industrial applications, the knowledge of the origin and the nature of the different excitations processes allows to understand the effects of the interaction between the electromagnetic wave and the matter. A part of the light energy is absorbed, a second part reflected and another part passes through the solid by inelastic scattering. These processes constitute the optical response of the material and can be characterized by the complex dielectric function which has an important role in the study of optical properties. This dielectric function is written in the complex formula [30]:
where ε1(ω) is the real part of the complex dielectric function, related to the polarization of the medium, and ε2(ω) is the imaginary part that characterized the absorption of the material.
The imaginary part ε2(ω) can be calculated from the momentum matrix element between the occupied and unoccupied wave functions in the selection rules, while the real part ε1(ω) of the complex dielectric function can be obtained using the Kramers–Kronig relation [31]. The imaginary and real parts of the dielectric function (ω) are given by:
An important parameter of this function ε1(ω) is the zero-frequency one ε1(0) called static dielectric constant (where only the electron contribution is taken into account) is related to the electron gap and explained by the Penn model [32, 33] given by the following expression:
where ħωpis the plasma energy; a small energy gap gives a large value of ε1(0).
All other optical constants such as absorption coefficient α(ω), refractive index n(ω), extinction coefficient k(ω), reflectivity R(ω), and energy loss L(ω) can be calculated using ε1(ω) and ε2(ω) by the following relations [34,35,36,37]:
We have calculated the optical properties of ZrSi3O8, ZrSiO4 and Zr3SiO8 compounds on the basis of our electronic structure calculations by GGA approximation for a photon of energy up to 13.5 eV.
3.3.1 Dielectric function ε(ω)
Figure 9 shows the evolution of the real ε1(ω) and imaginary ε2(ω) parts of the dielectric function of ZrSi3O8, ZrSiO4, and Zr3SiO8 compounds calculated using the GGA. We notice that we have two polarizations: ordinary (along the x and y direction) and extraordinary (along the Z direction). The average function is given by:

Real and imaginary part of the dielectric function of ZrSi3O8, ZrSiO4 and Zr3SiO8 using GGA approximation
The analysis of the ε2(ω) spectrum (Fig. 9b) shows that the first critical point occurs at about 1.78, 2.92 and 1.95 eV for ZrSi3O8, ZrSiO4 and Zr3SiO8, respectively. This point represents the threshold of direct transitions between the valence band maximum and the conduction band minimum Γv-Γc for ZrSi3O8 and indirect electronic transitions Γv-Mc for ZrSiO4 and Zr3SiO8 alloys; this is known as fundamental absorption threshold. This means that the electrical conductivity of these compounds can improve from high excitation energies. The most important contribution to this threshold can probably be attributed to inter-band transitions formed by electron transfer between occupied states, dominated by O-p states, and unoccupied states formed by hybridization of Si-p and Zr-d states.
The main peak, which reflects the absorption maximum, is located at 7.63, 7.27 and 6.03 eV for ZrSi3O8, ZrSiO4 and Zr3SiO8 compounds, respectively.
Figure 9a illustrates the variation of real part ε1(ω) of the dielectric function as a function of energy for the compounds ZrSi3O8, ZrSiO4 and Zr3SiO8. We noticed that for all these compounds the ε1(ω) function increases to maximum values at certain energy. After that, it gradually decreases and becomes negative at the following energy values: 10.65 eV (ZrSi3O8), 9.54 eV (ZrSiO4) and 7.91 eV (Zr3SiO8), where the dispersion at these energy values is zero and therefore the absorption is maximum. The values of the static dielectric constant ε1(0) are an important quantity, which is given by the lower limit ε1(ω → 0) of the compounds ZrSi3O8, ZrSiO4 and Zr3SiO8 are shown in Table 3.
To the best of our knowledge, there are no experimental data concerning these quantities in the scientific literature to be compared with our results. We find that the values of ε1(0) increase with decreasing energy gap Eg. This result can be explained by the Penn model [32, 33].
3.3.2 Absorption coefficient α(ω)
The linear absorption spectra of the compounds under study are shown in Fig. 10a. The absorption threshold, corresponding to the energy gap, starts at about 3.11 eV, 4.62 eV and 3.88 eV for ZrSi3O8, ZrSiO4 and Zr3SiO8, respectively. The absorption threshold is due to the electronic transitions from the occupied O-2p states located at the top of the valence bands to the empty Zr-d states that dominate the lower part of the conduction band. The first peak in the absorption spectrum occurs at 6.38 eV for ZrSi3O8, 5.63 eV for ZrSiO4 and 6.46 eV for Zr3SiO8. It is confirmed that these compounds are characterized by very low absorption in the visible region.

Variation of: a absorption coefficient α (ω), b refractive index n (ω),c extinction coefficient k (ω), d reflectivity R (ω), and e energy loss L (ω) as a function of energy for ZrSi3O8, ZrSiO4 and Zr3SiO8 using GGA approximation
3.3.3 Refraction index n(ω) and extinction coefficient k(ω)
The refractive index n(ω) and extinction coefficient k(ω) obtained for these compounds are plotted in Fig. 10b, c. From these figures we can see that the profile of these last quantities is almost similar for these compounds. In the visible range [1.59–3.26 eV] the refractive index of these compounds varies between (1.91 and 2.14) for ZrSi3O8 and (1.84–2.02) for ZrSiO4 and (2.06–2.36) for Zr3SiO8. In the ultraviolet region [3.26 − 12.4 eV] these spectra decrease with the increase in photon energy.
The value of static refractive index n (0) for ZrSi3O8, ZrSiO4 and Zr3SiO8 is 1.91, 1.87 and 2.02, respectively (show Table 3). The refractive index spectra show that the values of these compounds reach a maximum value of 2.39, 2.71 and 2.85 at energies 5.40, 5.07 and 4.23 eV, respectively.
The extinction coefficient K(ω) represents absorption phenomenon in the complex refractive index, it is directly related to the absorption coefficient. The extinction coefficient starts to increase from a threshold which is represented by the optical gap; this threshold is equal to 2.07 eV for ZrSi3O8 and 4.61, 3.79 for ZrSiO4 and Zr3SiO8 compounds, respectively.
3.3.4 Reflectivity R(ω) and electron energy loss function L(ω)
The reflectivity of light is important in the calculation of optical properties, Figs. 10d, 6e illustrate the optical reflectivity spectra R(ω) and the electron energy loss spectra L(ω) for ZrSi3O8, ZrSiO4 and Zr3SiO8, respectively. The analysis of the reflectivity spectra R(ω) shows that the reflectivity starts to increase from a low relatively value 9.53% for ZrSi3O8, 8.93% for ZrSiO4 and 11.71% for Zr3SiO8, in the visible region, all these compounds present a low reflectivity (less than 15%). Then, the reflectivity decreases rapidly for higher energies. This means that these substances behave like semiconductors. The static reflectivity coefficient R(0) for this materials is presented in Table 3.
The electron energy loss function L(ω) of a material is an important parameter describing the energy loss of a fast electron passing through the material. In this case, we represent the L(ω) for an interval of energy [0–13.5 eV]. The peaks appearing in the L(ω) spectrum represent a property associated with plasma resonance caused by a collective oscillation of valence electrons occurs at the plasma frequency \(\omega_{p}\) [38]. The important peaks of the L(ω) function are located at energies 10.05 eV for ZrSi3O8, 11.33 eV for ZrSiO4 and 13.05 eV for Zr3SiO8 corresponding to abrupt reductions in R(ω).
4 Conclusion
In summary, we have studied the optoelectronic properties of ZrxSi1-xO2 solid solutions (with x = 0−1) using the FP-LAPW method within the generalized gradient approximation (GGA) for the exchange correlation potential. The ground state of properties is in good agreement with the experimental and theoretical available results for ZrSiO4 compound. According to the electronic properties, ZrSiO4, Zr3SiO8 and ZrSi3O8 compounds show a semiconductor character. The ZrSiO4 and Zr3SiO8 compounds exhibit an indirect band gap of 4.565 eV and 3.675 eV, at Γ-M direction. However, ZrSi3O8 has a direct band gap of 2.008 eV at Γ-Γ direction. For optical properties, we have calculated the refraction index, absorption coefficient, extinction coefficient, reflectivity and energy loss for all our alloys. Based on these prediction results and compare it with available experimental and theoretical data, we can deduce that ZrSiO4 compound and its alloys Zr3SiO8 and ZrSi3O8 have a technological potential for diverse applications and this study can light the way for more theoretical and experimental investigation for these popular alloys for various chemical and physical applications in materials sciences.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
J. Zhang, Q. Zeng, A.R. Oganov, D. Dong, Y. Liu, Phys. Lett. A 378, 3549–3553 (2014)
K. Tanabe, T. Yamaguchi, Catal. Today 20(2), 185–197 (1994)
G. Orange, G. Fantozzi, F. Cambier, C. Leblud, M. Anseau, A. Leriche, J. Mater. Sci. 20, 2533–2540 (1985)
B.E. Burakov, J.M. Hanchar, M.V. Zamoryanskaya, V.M. Garbuzov, V.A. Zirlin, Radiochim. Acta 90(2), 95–97 (2002)
L. Kljajević, B. Matović, A. Radosavljević-Mihajlović, M. Rosić, S. Bosković, A. Devečerski, J. Alloys. Compd. 509(5), 2203–2215 (2011)
H. Hindam, D.P. Whittle, Oxide Met. 18, 245–284 (1982)
Z. Mutasim, C. Rimlinger, W. Brentnall, Am. Soc. Mech. Eng. GT-531, 1–10 (1997)
D.R. Clarke, C.G. Levi, Annu. Rev. Mater. Res. 33, 383–417 (2003)
G. Orange, G. Fantozzi, F. Cambier, C. Leblud, M. Anseau, A. Leriche, High tem-perature mechanical properties of reaction-sintered mullite/zirconia and mul-lite/alumina/zirconia composites. J. Mater. Sci. 20(7), 2533–2540 (1985)
J.F. Shackelford, W. Alexander, CRC Materials Science and Engineering Hand-book (CRC Press, Boca Raton, 2010)
D.C. Cranmer, Fibercoating and characterization. Am. Ceram. Soc. Bull. 68(2), 415–419 (1989)
J. Feng, D. Chen, W. Ni, S. Yang, Hu. Zijun, J. Non-Cryst. Solids 356, 480–483 (2010)
D.J. Cherniak, W.A. Lanford, F.J. Ryerson, Geochim. Cosmochim. Acta 55(6), 1663–1673 (1991)
W.J. Qi, R. Nieh, E. Dharmarajan, B.H. Lee, Y. Jeon, L. Kang, K. Onishi, J.C. Lee, Ultrathin zirconium silicate film with good thermal stability for alternative gatedielectric application. Appl. Phys. Lett. 77(11), 1704–1706 (2000)
A. Meldrum, L.A. Boatner, R.C. Ewing, Phys. Rev. Lett. 88, 025503–025506 (2001)
P. Blaha, K. Schwarz, G. Madsen, D. Kvasnicka, J. Luitz, Schwarz, wien2k, An augmented plane wave + local orbitals program for calculating crystal properties. (Vienna University of Technology, Austria, 2016)
J.P. Perdew, S. Burke, M. Ernzerhof, Phys. Rev. Lett. 77, 3865–3868 (1996)
F.D. Murnaghan, Proc. Natl. Acad. Sci. USA 30(9), 244–247 (1944)
B.A. Kolesov, C.A. Geiger, T. Armbruster, Eur. J. Miner. 13(5), 939–948 (2001)
M.B. Smirnov, S.V. Sukhomlinov, K.S. Smirnov, Phys. Rev. B 82, 094307 (2010)
R.W.G. Wyckoff, Crystal Structures, 2nd edn. (Interscience Publishers Inc, John Wiley & Sons, New York, 1965)
J.P. Crocombette, D. Ghaleb, J. Nucl. Mater. 257(3), 282–286 (1998)
M.S. Halati, H. Benchenane, M.H. Bouslama, A. Mokadem, A. Ouerdane, A. Bedjaoui, A. Bouhemadou, M.H. Guezzoul, B. Kharoubi, M. Abdelkrim, K.B. Bensassi, A. Baizid, Appl. Surf. Sci. 566, 150690 (2021)
K. Boudiaf, A. Bouhemadou, Y. Al-Douri, R. Khenata, S. Bin-Omran, N. Guechi, Electronic and thermoelectric properties of the layered BaFAg Ch (Ch = S, Se and Te): First-principles study. J. Alloys Compd. 759, 32–43 (2018)
G.M. Dongho Nguimdo, D.P. Joubert, Eur. Phys. J. B 88(5), 113 (2015)
A. Togo, I. Tanaka, Scr. Mater. 108, 1–5 (2015)
G. Kresse, J. Furthmüller, J. Hafner, Europhys. Lett. 32, 729 (1995)
P. Giannozzi, S. de Gironcoli, P. Pavone, S. Baroni, Phys. Rev. B 43, 7231 (1991)
T.T. Jiang, Q.Q. Sun, Y. Li, J.J. Guo, P. Zhou, S.J. Ding, D.W. Zhang, J. Phys. D Appl. Phys. 44(18), 185402 (2011)
C. Ambrosch-Draxl, J.O. Sofo, Comput. Phys. Commun. 175(1), 1–14 (2006)
Y. Ze-Jin, G. Yun-Dong, L. Jin, L. Jin-Chao, D. Wei, C. Xin-Lu, Y. Xiang-Dong, Chin. Phys. B 19(7), 077102 (2010)
D.R. Penn, Phys. Rev. 128(5), 2093 (1962)
A.H. Reshak, Z. Charifi, H. Baaziz, Eur. Phys. J. B 60, 463–468 (2007)
Z.E. Biskri, H. Rached, M. Bouchear, D. Rached, M.S. Aida, J. Electron. Mater. 45(10), 5082–5095 (2016)
X.D. Zhang, M.L. Guo, C.L. Liu, L.A. Zhang, W.Y. Zhang, Y.Q. Ding, Q. Wu, X. Feng, Eur. Phys. J. B 62, 417–421 (2008)
H. Rached, S. Bendaoudia, D. Rached, Mater. Chem. Phys. 193, 453–469 (2017)
A. Settouf, H. Rached, N. Benkhettou, D. Rached, Comput. Condens. Matter 19, e00377 (2019)
M. Fox, Optical properties of solids. Am. J. Phys. 70, 1269–1270 (2002)
Acknowledgements
This work was financially supported by DGRSDT (The general directorate for scientific research and technological development).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethical approval
I, the Corresponding Author, declare that this manuscript is original, has not been published before and is not currently being considered for publication elsewhere. I can confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. I further confirm that the order of authors listed in the manuscript has been approved by all of us. I understand that the Corresponding Author is the sole contact for the Editorial process and is responsible for communicating with the other authors about progress, submissions of revisions and final approval of proofs. Signed by the Corresponding Author on behalf of the all other authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ould Moussa, M., Kouidri, S., Rached, H. et al. Theoretical investigation on the optoelectronic properties of ZrxSi1-xO2 tetragonal hypothetical alloys from zircon family. Appl. Phys. A 128, 231 (2022). https://doi.org/10.1007/s00339-022-05362-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-022-05362-x