
Abstract
In this paper, polyvinyl alcohol/starch/graphene oxide nanocomposites containing glycerol were prepared via the solution-casting technique. The effect of different times (10, 20, 30, and 40 min) of nitrogen plasma irradiation on the structural, thermal, and dynamic mechanical properties of nanocomposites was studied. The dominancy of the chain scission process over crosslinking after irradiation for 10 min compared to the crosslinking at higher exposure times specifically at 30 min was proven by XRD. Also, field emission scanning electron microscopy confirmed these findings, where the most improved interface compatibility in nanocomposites shown after irradiation for 30 min. Thermogravimetric analysis indicated that after irradiation, the lowest thermal stability in nanocomposites shown in the sample irradiated for 10 min, while the highest thermal stability was shown in the sample irradiated for 30 min. It was found that the dynamic mechanical properties of nanocomposites were dependent on exposure time. The maximum storage modulus value was achieved after 30 min compared to the lowest value obtained after 10 min. Also, the calculated crosslinking density was increased by 49.8% after nanocomposite irradiation for 30 min compared to the unirradiated sample. Furthermore, the effect of plasma irradiation on both storage modulus and loss modulus of nanocomposites and their dependencies on temperature and frequency were studied.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The current worldwide attention of the environment boosts the researchers to replace petroleum-based plastic with eco-friendly materials such as biopolymers [1]. Among natural biopolymer, starch is extensively used due to its low cost, abundance, and biodegradability [2]. Starch consists mainly of amylose and amylopectine. Amylose is a linear molecule with a spiral structure, while amylopectine is a branched structure. The ratio of amylose to amylopectine controls the physical properties of starch [3, 4].
Unfortunately, the poor mechanical properties, high water sensitivity, and low processability limit the usage of starch in many applications, especially packaging purposes. So that, blending starch with other biopolymers such as polyvinyl alcohol (PVA) is required to overwhelm its drawbacks. PVA is biodegradable, highly polar soluble in water, highly mechanical strength, and non-toxic material. In contrast, PVA has some shortcomings such as high water uptake, high manufacturing cost, and weak dimensional stability. So that, the resulting mixture of blending PVA and starch provides higher mechanical properties, decreasing cost, a stable supporting medium, moisture resistance, rapid biodegradation. To further improve the physical properties of such blend the incorporation of filler such as graphene, [5] biochar [6], graphitic carbon nitride [7] can be applied. The addition of glycerol as a plasticizer in the PVA/starch blend advances those brittleness, flexibility, and processability [8, 9]. Graphene oxide (GO) nanofiller is usually used to improve the physical properties of polymers [10,11,12]. GO is rich with oxygen functional groups such as epoxy, carbonyl, and carboxyl which led to the construction of strong bonds between GO and starch [13]. The tensile strength and moisture resistance of PVA/starch blend containing glycerol were increased upon GO addition [2]. The irradiation of polymers and polymer nanocomposites is a well-known tool for modifying their properties [14].
The ionizing radiation such as electron beam [15], γ-rays [16], and ion-beam [17,18,19] was conducted to improve the mechanical, electrical, thermal properties of polymers [20]. Furthermore, gas plasma is a suitable technique that can effectively adjust the polymer’s surface properties for specific applications [21]. Plasma treatment can make changes in polymer’s structural, chemical, and physical properties like roughness to improve adhesion [22, 23] as well as creating well-defined surface nanostructures [24].
The initial processes which occur after interaction of a high-energy photon with an organic polymer are well established and do not depend basically on the substance’s chemical structure. However, these primary processes led to a cascade of secondary reactions which depends on the nature of the polymeric materials [25]. The polymer changes that depend on different factors such as the treatment period, as well as the power, temperature, density, and the frequency of plasma [26]. Plasma treatment of polymers with various gasses creates polar functional groups (–OH, C–O, and –COOH) on the polymer surface, which modify the surface energy of polymers and boosts attachments [27, 28]. It was reported that the nitrogen plasma treatment led to higher surface oxidation and produced a substantially higher concentration of polar functionalities in polymer surface compared with oxygen and argon plasma. As well as nitrogen plasma treatment rendered the polymer surface to be the most highly hydrophilic, biocompatible, and low surface roughness compared to oxygen and argon plasma [29, 30].
Figure 1 shows some works that have been made on starch/polyvinyl alcohol blend using other chemical and physical methods [31,32,33,34]. Also, the most published reports about PVA/starch/GO nanocomposites contain glycerol as a plasticizer lag behind the detailed study of the impact of nitrogen plasma on their dynamic mechanical properties. Motivated with this, the plasma irradiation was conducted to improve the thermal and dynamic mechanical properties of PVA/starch/GO nanocomposite to be used for packaging purposes. The plasma irradiation dose was controlled by change the time of irradiation. The physical properties of different irradiated samples have been studied compared to these unirradiated ones.

Some different chemical and physical processing of PVA/starch blend
2 Materials and methods
2.1 Materials
Graphite was gotten from Merk (Germany). H2SO4 (98%) and H3PO4 (85%) were procured from Sigma-Aldrich (USA). KMnO4, H2O2 (35%), ethanol (96%), HCl (30%), and NaOH were purchased from El Nasr Pharmaceutical chemicals company (Egypt). Polyvinyl alcohol (M. wt 75) was purchased from OCI (Seoul, Korea). Corn starch food-grade and glycerol were purchased from the Egyptian Starch & Glucose Manufacturing (Egypt).
2.2 Synthesis of graphene oxide
Graphene oxide (GO) material was synthesized according to the improved Hummer’s method [35]. Briefly, 1 gram of graphite was added to 100 ml solution mixture from H2SO4 + H3PO4 (3:1) in an ice bath. Then, 6 gram of KMnO4 was added slowly to the above mixture which was kept under continuous stirring for about 24 h. Deionized water was then further added which changed the color of the mixture from the dark purplish-green to the dark brown. To stop the oxidation reaction, 30 ml of H2O2 solution was added to mixtures. The obtained graphite oxide solid was washed with 1 M HCl aqueous solution and repeatedly washed with deionized water until a pH reaches up to 3.
2.3 Preparation of PVA/starch/GO nanocomposite
The solution casting technique was used to fabricate PVA/starch/GO film. First, dissolving of 1 gram from PVA in distilled water (30 mL) was made by heating at 90 °C for 30 min. Similarly, 1 gram of starch dissolved separately, in distilled water (20 ml) and heated to 90 °C until gelatinization. Then, the starch and PVA solutions were mixed in a ratio of 1:1. Then, 1 ml of GO solution (GO dissolved in distilled water with ratio of 1:1) was added to the above solution followed by stirring for about 15 min. Then, 1 ml glycerol plasticizer was added. The composite was then stirred for ~ 30 min. Lastly, the suspensions were transferred onto a Petri dish for drying at ambient temperature for 72 h.
2.4 Plasma irradiation of PVA/starch/GO nanocomposites
Figure 2 shows the schematic representation of the cold plasma reactor used for samples irradiation. It consists of a stainless steel cylindrical anode of 40 mm diameter, 60 mm length and stainless steel disk cathode of 60 mm outer diameter and 5 mm inner aperture diameter.

Schematic representation of cold plasma source
The anode and the cathode are insulated by Teflon of thickness 4 mm. The distance between a collector plate to the exit aperture was 4 mm to measure the output plasma current. The samples were irradiated for different times of 10, 20, 30, and 40 min using nitrogen plasma with energy of 2 keV. The current density (Ip) was 1 mA/cm2, the discharge current (Id) was 3 mA, the working pressure was 1.2 × 10− 3 mbar, and ion flux was 1.25 × 1016 ion/cm2 sec.
2.5 Characterization techniques
The structural analysis of nanocomposites was conducted by X-ray diffractometer (XRD, Shimadzu) using CuKα source (λ = 1.5405A°). The scan was done at 4–90° in continuous mode. The generator voltage and current were 40 kV and 30 mA, respectively. The chemical structure of composites was investigated using Fourier transform infrared spectroscopy FT-IR (Shimadzu Prestige-21 Spectrophotometer) in the 4000–400 cm−1 range. The surface morphology of samples was examined by field emission scanning electron microscope (FESEM Sigma 300 VP, Carl Zeiss, Germany). The samples were sputter-coated at 30 mA for 30 s by a gold thin layer. The thermal stability of the samples is tested via thermogravimetric analysis (TGA; Shimadzu -50) in the temperature range of 25–600 °C. The heating was performed at the rate of 10 °C min−1 under a nitrogen atmosphere. The dynamic mechanical properties of nanocomposites were tested utilizing dynamic mechanical analysis (Triton Instruments). Rectangular specimens were tested at different frequencies (0.5, 1, 2, 3 Hz) in the tension mode from room temperature to 160 °C with a heating rate of 10 °C min−1.
3 Results and discussion
3.1 Structural analysis
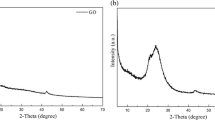
Figure 3 shows the XRD pattern of pure PVA, pristine starch, and GO. Pure PVA shows a strong broad peak at 19.9° analogous to the (1 1 0) reflection plane. The observed shoulder peak at 22.9° as well as a peak at 40.8° ascribed to the (2 0 0) and (111) planes, respectively [36, 37]. Pristine starch shows three peaks, the first at 23.25° distinguishing the B-type polymorphs which usually existing in tuber starch. The second peak at 14.93° corresponds to A-type polymorphs which are chiefly associated with the cereal starch. The third peak at 17.76° characteristic of both A- and B-type polymorphs [38, 39]. GO displays a strong peak located at 9.6° with d = 9.2 Å analogous to the (001) plane which characterizes graphitic layers rich in oxygen functional groups [40].

XRD pattern of pure PVA, neat starch, and GO
XRD patterns of unirradiated and irradiated nanocomposites are presented in Fig. 4. Compared to pure PVA and neat starch, the unirradiated nanocomposite sample shows broad peaks at 19.6° and 40.8°. The absence of starch peaks in nanocomposites pattern due to the crystalline structure damage of the pristine starch powder caused by gelatinization during the blending process [41].

XRD pattern of unirradiated and irradiated PVA/starch/GO nanocomposites
The decreased intensity of GO peak after incorporation into nanocomposite indicates the strong interaction and homogeneous dispersion of GO sheets in the polymers matrix [13, 37]. This behavior is different from other reported results where the XRD pattern of PVA was dominant in PVA/starch blend containing glycerol [42]. The dramatic decreasing intensity of the main diffraction peak of PVA in nanocomposite demonstrates the decrease in PVA crystallinity. The reduction of polymer crystallinity after incorporation of GO is due to reduction polymer chain mobility owing to the formation of a bounded polymer region. Such region positioned around filler surface nearby the interface region displays dissimilar characteristics compared to the pristine polymer due to the interfacial interactions between the polymer host matrix and the filler surface. Also, the mobility of the polymer chains inside this region is significantly hindered, and the filler content influences the degree of mobility constraint polymer chains [43]. Thus, it can be say that the interactions between oxygen groups of GO and the –OH of PVA constrain the polymer’s motion causing a reduction in its crystallinity [37].
It can be seen that the irradiation not only causes the upsurge in the main PVA peak intensity but the broadening of the diffraction peak. If the diffraction peak broadens, it can be anticipated that the mean crystallite size will decline [44]. The crystallite size lessening is induced by crosslinking on the lamellae surfaces [44, 45]. So, the remarkable dominance of the main PVA peak after irradiation with 10 min reveals chain scission process dominates the crosslinking. In contrast, an increase in the amorphous phase due to the dominancy of molecular chain crosslinking of nanocomposite over the chain scission at longer irradiation times observed specifically at 30 min [46].
As shown in Fig. 5, FTIR spectra of pure PVA, neat starch and GO show the absorption bands at ~ 3221, 3277, and 3190 cm−1, respectively, ascribed to the stretching vibration of the OH group inside them [47]. The basic band of pure PVA at 2914 cm−1 represents the alkyl (C–H) stretching and the band at 1724 cm−1 due to the C=O group. The absorption bands at 1428–1241 cm−1 ascribed to C–H wagging in pure PVA. The bands at 1085 and 1025 cm−1 assigned the vibration of C–O in C–OH groups, while bands at 944 and 846 cm−1 correspond to C–O and C–C vibrations, respectively [48, 49].

FTIR spectra of pure PVA, pristine starch, and GO
The neat starch shows the C–H stretching vibration of aliphatic groups at 2927 cm−1, adsorbed water at 1631 cm−1, and C–H bending for alkane vibration at 1341 cm−1. Also, C–C and C–O stretching at 1148 cm−1 and C–O–H bending vibrations at 1002 cm−1 were detected [8]. The spectrum of GO shows abundant oxygen functional groups stretching vibration of C=O at 1720 cm−1, C–OH at 1395 cm−1, C–O at 1210- 1056 cm−1, and C=C a 1623 cm−1 [50].
As shown in Fig. 6, the incorporation of starch and GO into PVA results in the OH, and C–H stretching vibrations of neat PVA were shifted to a higher frequency and became stronger. Also, decreased intensity of C-H and C–C bands at 1724 and 840 cm−1 recommends the formation of chemical conjugation of starch with PVA molecules. The almost disappearance of bands at 1370 and 1428 cm−1 indicates the decoupling between O–H and C–H vibrations due to the bonding interaction with O–H and both starch & GO [51]. The appearance of the band at 1655 cm−1 and redshift of the C–O band from 1085 cm−1 to 1034 cm−1 indicates strong interaction occurred between PVA, starch, and GO [51]. The intensity of bands at 3379, 2928, 1250, 1727, and 1034 cm−1 has been changed with varying plasma irradiation time indicates the rearrangement of PVA chains by nitrogen ions irradiation[51], where the lower intensity of bands at 3379, 1034, and 1424 cm−1 was observed after 10 min from irradiation and then it increased and reached its highest value after 30 min of exposure. For example, the intensity of absorption peak at 1034 cm−1 was ≈ 10, 9, 16 and 13% corresponding to irradiation time of zero, 10, 20, 30, and 40 min, respectively. The decreased intensity of these bands suggests the degradation of the sample after 10 min from plasma exposure, while the sample crosslinking occurs after lengthen the exposure time and reaches its maximum after 30 min [52, 53]. The intensity of OH groups decreased after irradiation for 10 min then increased to its maxima at 30 min. This behavior assigned to the chain scission at 10 min causes the production of both oxygen and hydrogen radicals. With increasing irradiation time, the oxygen radicals can interact with hydrogen radicals or with bonded hydrogen to form OH groups [53].

FTIR spectra of unirradiated and irradiated PVA/ starch/GO nanocomposites
The surface and cross section morphologies of unirradiated and irradiated PVA/starch/GO blends were depicted in Fig. 7. From Fig. 7a–e, the rough surface of unirradiated blend with many globules due to immiscibility and the coexistence of both PVA and starch phases [2].

FESEM surface area images of PVA/starch/GO nanocomposite unirradiated (a) irradiated with 10 (b), 20 (c), 30 (d), and 40 (e) minutes of nitrogen plasma
After plasma irradiation, the size of surface globules change and the most improved interface compatibility for nanocomposites showed after 30 min. This behavior reflects the radiation reorganization and crosslinking [54]. By further increasing irradiation time to 40 min, the size of the globules was increased again which may designate prevail the chains scission process over the crosslinking in the polymer matrix [15].
3.2 Thermal properties
The thermogravimetric analysis of PVA/starch/GO films was performed to understand the impact of nitrogen plasma on the thermal stability of nanocomposites.
Figure 8 shows the TGA curves for PVA/starch/GO films irradiated with different times of nitrogen plasma compared to unirradiated ones. For the unirradiated nanocomposite, the first major weight loss happens in the temperature range 40–110 °C owed to the water elimination. The second weight loss happens in temperature span 140–247 °C attributed to the removal of water and the volatile components such as glycerol. The third weight loss temperature span is 274–330 °C owing to the thermal degradation of sample intermolecular hydrogen bonding. The fourth weight loss happens between 375 and 445 °C corresponds to the decomposition of the sample’s main backbone chain. While the last weight loss occurs in temperature span 460–516 °C due to the by-products released during extensive degradation of polymer nanocomposite backbone [55].

TGA curves of unirradiated and irradiated PVA/starch/GO nanocomposite
The thermal stability of nanocomposites decreased after exposure to plasma for 10 min. By further increasing exposure time, the thermal stability begins to increase again till the thermal stability of nanocomposites irradiated with 30 min surpasses the unirradiated nanocomposite in temperature span above 270 °C. By increasing the exposure time to 40 min the thermal stability decreased again. The remaining weight of unirradiated sample was 1.13% compared to 0.411%, 0.91%, 2.3%, and 0% for those irradiated for 10, 20, 30, and 40 min, respectively. The effect of plasma on the thermal stability of nanocomposites could be ascribed to the competitive process of plasma radiation-induced crosslinking and degradation reaction occurred concurrently [54]. Based on this argument, the TGA results indicate dominancy of nanocomposites degradation at 10 min of irradiation, while the crosslinking dominancy recovered by increased exposure time and reach its maximum at 30 min. These findings are consistent with XRD, FTIR, and FESEM results.
3.3 Dynamic mechanical properties
3.3.1 The storage and loss modulus
The dynamic mechanical analysis gives strong information about the phase structure and viscoelastic properties of nanocomposites. The impact of temperature on the storage modulus and loss modulus at 1 Hz for unirradiated and irradiated PVA/starch/GO nanocomposite is shown in Fig. 9a, b. The storage modulus reduced as temperature increased and had three separate zones. The first was a glassy zone with a high module attributed to the reduction of the chains' mobility. The second is the transition zone with a modulus decrease which shows the temperatures of glass transition. The third is the rubber zone with small modulus values, attributed to the high mobility of polymer chains, attributed to energy dissipation. [56]. The storage modulus of PVA/starch/GO nanocomposite at 30 °C was significantly dropped from 6.3 to 4.5 MPa (approximately 27.5%, decreasing) after irradiation for 10 min. Then, with increasing irradiation time to 20 min, 30 min, and 40 min, the storage modulus was increased to 5.9 MPa, 9.4 MPa, and 8.2 MPa, respectively. The maximum modulus was at irradiation of 30 min representing ~ 49% modulus enhancement compared to unirradiated nanocomposite. Also, the loss modulus at 30 °C was decreased from 0.74 to 0.33 MPa after 10 min. With further increasing exposure time to 20, 30, and 40 min, the loss modulus increased to 0.49 MPa, 0.93 MPa, and 1.02 MPa, respectively.

a Storage modulus and b loss modulus dependency on temperature for the unirradiated and irradiated nanocomposites
As described early, two phenomena can occur concomitantly when polymer composites were subjected to irradiation, namely crosslinking and chain degradation. In the first phenomenon, a combination reaction stabilizes free radicals and results in crosslinking between the molecules, increasing the modulus. Inversely, the second process leads to break the backbone of the chain at a higher dose. Polymers are then degraded into small fragments, causing a decrease in the modulus [57, 58]. In both phenomena, the exposure time greatly affects the degree of crosslinking or chain degradation.
To further verify the influence of exposure time on the degree of crosslinking and consecutively on mechanical properties of PVA/starch/GO nanocomposite, polymer's crosslink density was calculated according to the elasticity theory using the following equation: [59, 60]
where υ is the crosslinking density, E′ is the storage modulus in the rubbery region, while R (8.31 J⋅K−1⋅mol−1) is the gas constant, and T is the absolute temperature of (Tg + 50) K. The calculated degree of crosslinking density is presented in Fig. 10. The outcomes display that nanocomposite irradiation for 10 min and 20 min reduced the crosslinking by 10.4% and 0.8% compared with unirradiated composite, respectively, leading to a decrease in storage modulus as shown in Fig. 7 a. Dissimilar at exposure time’s 30 min and 40 min, the crosslinking increased by 49.8% and 8.6% compared with unirradiated composite, respectively. This causes’ a significant improvement in storage modulus as described earlier. So that, the best exposure time considering the maximum crosslinking achieved was 30 min. The decreasing crosslinking after 30 min ascribed to the predominance of chain scission process caused by irradiation over the crosslinking as described earlier.

The crosslinking for the unirradiated and irradiated nanocomposites
3.3.2 The effect of frequency
The variation of storage modulus with frequency as a function of the temperature of unirradiated and different irradiated PVA/starch/GO nanocomposites is shown in Fig. 11a, b.

The frequency dependency of the storage modulus of a unirradiated and b irradiated PVA/starch/GO nanocomposite
The storage modulus is significantly affected by frequency especially at high temperatures. The modulus values were dropped at temperature ~ 60 °C and continue steadily till a temperature of 120 °C. The change in dynamic mechanical properties is accompanying with the creation of microscopic cracks and the breaking down of crosslinking bonds [61]. At high temperatures, usually breaking down of the filler agglomerates and the breaking down of the bonds between the filler and polymer matrix have been occurred, leading to a significant change in modulus values.
It was also found that the storage modulus curves shift to higher temperatures with an increase in frequency. This is because when the material is subjected to constant stress, the modulus will be declined over a period owed to the material subject to molecular rearrangement to minimize the localized stresses. Modulus measurements performed over a short time (high frequency) result in higher values, whereas measurements were taken over long times (low frequency) result in lower values [61].
The relation between glass transition temperature and the frequencies for the unirradiated and irradiated PVA/starch/GO nanocomposite is shown in Fig. 12. The impact of frequency on the glass transition (Tg) of the nanocomposites can be elucidated by the Arrehenius relationship. At the glass transition region, the activation energy (E) for the relaxation process is proportional to the slope of a plot of frequency (the natural log) and the reciprocal of T (K). The activation energy (E) can be calculated from the slope of the regression line by the following equation [62]:
where F is the frequency, R is the universal gas constant and Tg is the absolute glass transition temperature. Table 1 presents measured E values and the coefficient of the fitting curves (r2). The energy of activation is aligned to the energy needed to encourage the initial movement of polymer chain molecular segments [63]. The highest activation energy was obtained at an exposure time of 30 min (≈ 944.6 kJ/mol). The higher restriction of polymer chains mobility and the higher activation energy of the nanocomposites designate the existence of high values of crosslinking. This result is consistent with the previous findings.

The variation of ln (F) with Tg for the unirradiated and irradiated PVA/starch/GO nanocomposite
4 Conclusion
PVA/starch/GO nanocomposite was successfully prepared by the solution casting method. The influence of different times of nitrogen plasma irradiation on nanocomposite was studied. Compared to the broad peak appearance of PVA main peak in XRD pattern of unirradiated composite, the remarkably predominance of this peak was observed after plasma exposure for 10 min; then, the increased amorphous phase was observed with increased exposure times. This behavior exhibited the dominancy of the chain scission process over crosslinking after 10 min of irradiation compared to prevailing crosslinking at higher exposure times specifically at 30 min. This result confirmed with FTIR where the decreased intensity of bands located at 3379, 1034, and 1424 cm−1 observed after 10 min from irradiation and then it increased and reached its highest intensity value after 30 min of exposure. Also, FESEM confirmed these findings, where the most improved interface compatibility in PVA/starch/GO showed after plasma irradiation for 30 min. The thermal properties of nanocomposites were influenced by plasma irradiation time, where the worst thermal stability of nanocomposites was showed after 10 min, whereas the best thermal stability was displayed after 30 min. The storage modulus of unirradiated composites was dropped upon irradiation for 10 min from 6.3 to 4.5 MPa. With further increasing exposure time, the storage modulus of nanocomposites enhanced to its highest value after 30 min. Similar behavior of variation of loss modulus with irradiation time was observed which reflects the effect of plasma irradiation time on the crosslinking of nanocomposites degree. The calculated crosslinking density of nanocomposite at different irradiation times proved this behavior. Furthermore, the highest calculated activation energy was 944.6 kJ/mol after irradiation for 30 min. This reflects a higher crosslinking degree by upsurges the mobility restraint of the polymer chains.
References
W. Wang, H. Zhang, Y. Dai, H. Hou, H. Dong, Iran. Polym. J. 24, 687–696 (2015)
Z. Wu, Y. Huang, L. Xiao, D. Lin, Y. Yang, H. Wang, Y. Yang, D. Wu, H. Chen, Q. Zhang, Int. J. Biol. Macromol. 123, 569–575 (2019)
L. Avérous, P.J. Halley, Biofuel. Bioprod. Bior. 3, 329–343 (2009)
F. Zia, K.M. Zia, M. Zuber, S. Kamal, N. Aslam, Carbohydr. polym. 134, 784–798 (2015)
O.A. Bin-Dahman, M. Rahaman, D. Khastgir, M.A. Al-Harthi, Can. J. Chem. Eng. 96, 903–911 (2018)
P. Terzioğlu, F.N. Parin, J. Nat. Appl. Sci. 24(1) (2020)
A. Ahmed, M.B.K. Niazi, Z. Jahan, G. Samin, E. Pervaiz, A. Hussain, M.T. Mehran, J. Polym. Environ. 28, 100–115 (2020)
N.A. Maziad, A.B.E.D.A. El, S.E. Hamouly, M.A.E.A.M. El, S.A. Rizk, N.R. Nasef, J. Biosci. Med. 6, 33–56 (2018)
T.J. Gutiérrez, N.J. Morales, E. Pérez, M.S. Tapia, L. Famá, Food. Packag. Shelf Life. 3, 1–8 (2015)
Y. Zhang, Q. Zhang, D. Hou, J. Zhang, Appl. Surf. Sci. 504, 144152 (2020)
Y. Xie, C. Liu, W. Liu, L. Liang, S. Wang, F. Zhang, H. Shi, M. Yang, Colloids Surf A: Physicoch. Eng. Asp. 124627 (2020)
J. Osicka, M. Mrlik, M. Ilcikova, I. Krupa, P. Sobolčiak, T. Plachý, J. Mosnáček, React. Func. Polym. 148, 104487 (2020)
E. Sedaghat, A.A. Rostami, M. Ghaemy, A. Rostami, Characterization. J. Therm. Anal. Calorim. 136, 759–769 (2019)
R. Ramola, S. Chandra, A. Negi, J. Rana, S. Annapoorni, R. Sonkawade, P. Kulriya, A. Srivastava, Phys. B: Condens. Matter 404, 26–30 (2009)
S.-T. Bee, L.T. Sin, C. Ratnam, B.-F. Yap, A. Rahmat, Nucl. Instrum. Methods. Phys. Res. Secti B: Beam Inter Mater Atom. 416, 73–88 (2018)
A. Akhavan, F. Khoylou, E. Ataeivarjovi, Radiat. Phys. Chem. 138, 49–53 (2017)
M. Ghobashy, A.A. Reheem, N. Mazied, Int. Polym. Process. 32, 174–182 (2017)
A.A. Reheem, A. Atta, T. Afify, Surf. Rev. Lett. 24, 1750038 (2017)
A. El-Saftawy, A.A. Reheem, S. Kandil, S. Abd El Aal, S. Salama, Appl. Surf. Sci. 371, 596–606 (2016)
M. Zhai, F. Yoshii, T. Kume, Radiation modification of starch-based plastic sheets. Carbohydr. polym. 52, 311–317 (2003)
A. Hassan, S. Abd El Aal, M. Shehata, A. El-Saftawy, Surf. Rev. Lett. 26, 1850220 (2019)
J. Dewez, E. Humbeek, E. Everaert, A. Doren, P. Rouxhet, Polym. Solid Interfc. 463–474 (1991)
S. Wu, Polymer Interfacial and Adhesion, 1st edn. (Marcel Dekker Inc., New York, 1982)
K. Ostrikov, Plasma nanoscience: basic concepts and applications of deterministic nanofabrication, (John Wiley & Sons, 2008)
S. Dorey, F. Gaston, S.R. Marque, B. Bortolotti, N. Dupuy, Appl. Surf. Sci. 427, 966–972 (2018)
A. Bismarck, J. Springer, Wettability of Materials: Plasma Treatment Effects Encyclopedia of Surface and Colloid Science., 2nd edn. (Taylor & Francis, 2006) pp. 6592–6610
A. Vesel, I. Junkar, U. Cvelbar, J. Kovac, M. Mozetic, Interfaces Thin films 40, 1444–1453 (2008)
A. Vesel, M. Mozetic, Vacuum 86, 634–637 (2012)
I. Junkar, A. Vesel, U. Cvelbar, M. Mozetic, S. Strnad, Vacuum 84, 83–85 (2010)
S. Roy, C.Y. Yue, Plasma Processes Polym. 8(5), 432–443 (2011)
D. Domene-López, M.M. Guillén, I. Martin-Gullon, J.C. García-Quesada, M.G. Montalbán, Carbohydr. Polym. 202, 299–305 (2018)
P. Yin, X. Dong, W. Zhou, D. Zha, J. Xu, B. Guo, P. Li, RSC Adv. 10(40), 23632–23643 (2010)
G.V. Ravindra, S.K. Vineeth, P.T. Gadekar, J. Mater Environ. Sci. 11, 704–712 (2020)
N.C. Dafader, N. Rahman, M.F. Alam, M.S. Rahaman, M.A. Rahim, A.H.M. Kamal, Polym. Res. J. 11(3), 121–132 (2017)
D.C. Marcano, D.V. Kosynkin, J.M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L.B. Alemany, W. Lu, J.M. Tour, ACS Nano. 4, 4806–4814 (2010)
W.H. Eisa, Y.K. Abdel-Moneam, Y. Shaaban, A.A. Abdel-Fattah, A.M.A. Zeid, Mater. Chem. Phys. 128, 109–113 (2011)
M. Cobos, M.J. Fernández, M.D. Fernández, Nanomaterials 8(12), 1013 (2018)
M. Shahbazi, G. Rajabzadeh, S. Sotoodeh, Int. J. Biol. macromol. 104, 597–605 (2017)
M.-C. Popescu, B.-I. Dogaru, M. Goanta, D. Timpu, Int. J. Biol. macromol. 116, 385–393 (2018)
A. Buchsteiner, A. Lerf, J. Pieper, J. Phys. Chem. B. 110, 22328–22338 (2006)
P. Boonsuk, A. Sukolrat, K. Kaewtatip, S. Chantarak, A. Kelarakis, C. Chaibundit, J. Appl. Polym. Sci. 137(26), 48848 (2020)
J. Jose, M.A. Al‐Harthi, M.A.A. AlMa'adeed, J. Bhadra Dakua, S.K. De, J. App. Polym. Sci. (2015)
H.-D. Huang, J.-Z. Xu, Y. Fan, L. Xu, Z.-M. Li, J. Phys. Chem B. 117, 10641–10651 (2013)
Z. Zhudi, Y. Wenxue, C. Xinfang, Radiat. Phys. Chem. 65, 173–176 (2002)
A. Gupta, R. Bajpai, J. Keller, J. Polym. Res. 15, 275–283 (2008)
M. Shehata, S. Radwan, H. Saleh, Z. Ali, J. Radioanal. Nucl. Chem. 323, 897–902 (2020)
R. Kizil, J. Irudayaraj, K. Seetharaman, J. Agric. Food. chem. 50(14), 3912–3918 (2002)
H.M. Afzal, F. Shehzad, M. Zubair, O.Y. Bakather, M.A. Al-Harthi, J. Therm. Anal. Calorim. 139(1), 353–365 (2020)
H. Tiana, J. Yan, A.V. Rajulu, A. Xiang, X. Luo, Int. J. Biol. Macromol. 96, 518–523 (2017)
K.N. Kudin, B. Ozbas, H.C. Schniepp, R.K. Prud’Homme, I.A. Aksay, R. Car, Nano lett. 8, 36–41 (2008)
R.P. Chahal, S. Mahendia, A. Tomar, S. Kumar, J. Alloy. Compd. 538, 212–219 (2012)
A. Al-Sabagh, E. Taha, U. Kandil, G.A. Nasr, M.R. Taha, Nanomaterials 6, 169 (2016)
R. Huszank, E. Szilágyi, Z. Szoboszlai, Z. Szikszai, Nucl. Instrum. Methods. Phys. Res. Secti B: Beam Inter Mater Atom 450, 364–368 (2019)
D. Guo, Radiat. Phys. Chem. 130, 202–207 (2017)
W. Rahman, L.T. Sin, A. Rahmat, A. Samad, Carbohydr. polym. 81, 805–810 (2010)
K.P. Menard, N. Menard, in Encyclopedia of Analytical Chemistry: Applications, Theory and Instrumentation (2006), pp. 1–25
K. Motaleb, R. Milašius, A. Ahad, Fibers 8, 58 (2020)
E. Manaila, M.D. Stelescu, G. Craciun, in Advanced Elastomers—Technology, Properties and Applications (2012), pp. 3–34
A. Al-Sabagh, E. Taha, U. Kandil, A. Awadallah, G.A.M. Nasr, M.R. Taha, Polym. 9, 94 (2017)
L.E. Nielsen, J. Macromol. Sci. C. 3, 69–103 (1969)
T. Murayam, (Elsevier Scientific Pub. Co., 1982)
W.K. Goertzen, M. Kessler, Compos. B: Eng. 38, 1–9 (2007)
H.L. Ornaghi Jr., A.S. Bolner, R. Fiorio, A.J. Zattera, S.C. Amico, J. App. Polym. Sci. 118, 887–896 (2010)
Acknowledgements
The authors would like to express their gratitude and appreciation to National Center for Radiation Researches and Technology (NCRRT), Egyptian Atomic Energy Authority, and Polymer Nanocomposite Center of Excellence (Project ID: 37200), Egyptian Petroleum Research Institute (EPRI), for supporting and providing materials for this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Atta, M.M., Taha, E.O. & Abdelreheem, A.M. Nitrogen plasma effect on the structural, thermal, and dynamic mechanical properties of PVA/starch/graphene oxide nanocomposite. Appl. Phys. A 127, 532 (2021). https://doi.org/10.1007/s00339-021-04671-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-021-04671-x