
Abstract
We report the preparation of 0.1% Eu3+; x% Gd3+ (0 ≤ x ≤ 1.2) ZnAl2O4 phosphors using the citrate sol–gel method. X-ray diffraction (XRD) data revealed that all annealed samples consisted of the single phase of cubic ZnAl2O4 structure. The scanning electron microscopic (SEM) images indicated pronounced effect of dual doping on the surface morphology of the phosphor. The estimated crystal sizes estimated by the XRD was confirmed by the high-resolution transmission electron microscopy (HR-TEM) to be approximately around 20 nm. The photoluminescence (PL) spectroscopy results revealed four distinct emission peaks located at 393, 400, 578 and 618 nm. The peaks at around 393 and 400 nm were attributed to the defect levels located at different positions on the host material (ZnAl2O4). The emission peak at 578 and 618 nm were attributed to the 5D0 → 7F1 and 2D0 → 7F2 characteristic transition within the Eu3+ ions. The International Commission on Illumination (CIE) colour coordinates revealed that the emission colour was not influenced by varying the concentration of Gd3+.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Zinc aluminate (ZnAl2O4) is one of the representative compounds in the family of metal aluminate, with the spinel structure, and it is widely used as ceramic, electronic, catalytic material, etc. [1]. ZnAl2O4 also possesses a remarkable combination of properties such as high thermal, chemical stability, better ductility, low surface acidity, moderate conductivity and high quantum yields [1,2,3]. ZnAl2O4 is a wide band gap semiconductor with an optical band gap of ~ 3.8 eV, which makes it useful for photo-electronic devices [2]. Recently, researchers have also shown that ZnAl2O4 phosphor has many applications in stress imaging devices, thin film electroluminescent displays and mechano-optical stress sensors [2, 3]. ZnAl2O4 is known to crystalize under the normal spinel structural configuration with the chemical formula AB2O4, where A represents the divalent cation (Zn2+) that occupies the tetrahedral sites and B represents the trivalent cation (Al3+) that occupies the octahedral sites of a close packed crystal structure that belongs to the space group Fd\(\stackrel{-}{3}\)m. The unit cell contains 32 oxygen (O2−) anions, 16 Al3+ cations and 8 Zn2+ cations [4]. The literature has shown that ZnAl2O4 can be synthesized by numerous types of wet chemical techniques [5, 6], e.g. to fabricate ZnAl2O4 doped with rare earth (RE) metals [6]. Strek et al. [7] prepared ZnAl2O4:Eu3+ via the hydrothermal method and used X-ray diffraction (XRD) to demonstrate the formation of a single phase of ZnAl2O4 nanopowders with high crystallinity. The size of the nanocrystallites was determined to be 8 and 15 nm for samples heated at 500 and 1000 °C, respectively. However, details of the synthesis and spectroscopic characterization of Gd-doped ZnAl2O4 nanophosphor were not located in open resources. Gd3+ has been reported as part of the host [8] and as a dopant of Y2O3 [9]. Zatsepin et al. [8] studied the intrinsic luminescence of Gd2O3. They reported that the cubic polymorph exhibited an intrinsic luminescence due to “defective” Gd3+ ions at 315 nm. This ultraviolet emission is the consequence of 232 and 275 nm 4–4f excitation. Tamrakar et al. [9] investigated the emission of cubic Y2O3:Gd3+. The photoluminescence (PL) spectra suggested the presence of three distinct emission peaks located at 314, 317 and 395 nm. These peaks were due to the transitions between excited states 6Ij, 6Pj to the ground energy level 8S7/2 of Gd3+ ions. Eu3+ has previously been reported as a dopant in ZnAl2O4 [10] and other host materials [11]. Motloung et al. [10] studied the ZnAl2O4:0.1% Ce3+;x% Eu3+ dual-doped system and the PL results showed emission peaks that were attributed to the host defects and Ce3+ and Eu3+ transitions, as well as confirming energy transfer (ET) from Ce3+ → Eu3+. On the contrary, Chengaiah et al. [11] showed the existence of energy transfer from Eu3+ → Ce3+ in Na3Gd(PO4)2:Eu3+; Ce3+ co-doped phosphor. Thus, the ET pathways or direction can be controlled to either favour Ce3+ → Eu3+ or Eu3+ → Ce3+.
From an in-depth literature review, it is clear that there are many investigations on the doping of ZnAl2O4 with different ions [5,6,7, 10]. However, there is no reported investigation exploring the ZnAl2O4 nano-sized host co-doped with Eu3+ and Gd3+ ions. The present report is based on an investigation pertaining to the effect of varying the Gd3+ concentration on the structure and optical properties of ZnAl2O4:0.1% Eu3+; x% Gd3+ co-doped phosphor material synthesized by the citrate sol–gel process. The primary objective of the current work is to develop alternative phosphor materials for practical applications such as for light emitting diodes (LEDs). The photophysics related to the excitation and de-excitation processes is explained in detail.
2 Experimental procedure
2.1 Samples synthesis
ZnAl2O4 (host), singly doped (with 0.1% Eu3+ or 0.1% Gd3+) and co-doped with 0.1% Eu3+; x% Gd3+ (0 ≤ x ≤ 1.2), nanophosphors were synthesized using the citrate sol–gel technique. The host (undoped) material was prepared by dissolving Zn(NO3)2·6H2O (98%), Al(NO3)3·9H2O (98.5%) and citric acid (CA) C8H8O7·H2O (99%) in deionized water. The stoichiometric molar ratio of Zn:Al and Zn:CA was kept at 1:2 and 1:0.75, respectively. Specified amounts of Gd(NO3)3·6H2O (99.9%) and Eu(NO3)3·5H2O (99.9%) were added into separate beakers to singly dope with 0.1% Eu3+ and 0.1% Gd3+. All chemicals were purchased from Sigma-Aldrich. The co-doped samples were prepared by keeping the 0.1% Eu3+ concentration constant and varying the x% Gd3+ concentration over a range 0 ≤ x ≤ 1.2. The heating temperature was kept at ∼80 °C while constantly stirring using a magnetic stirrer until gels were formed. These were subsequently annealed in air at 800 °C in a furnace for 1 h. The products were crushed using a pestle and mortar to produce powders.
2.2 Characterization
The crushed powder samples were then analysed using different techniques. The crystal structure and phase composition of the prepared powder samples were analysed with a Bruker D8-Advance powder with a CuKα (1.5405 Å) radiation. The prepared powder was carbon coated using the Quorum carbon coater and the Tescan VEGA3 scanning electron microscope (SEM) coupled with Oxford XMaxN energy-dispersive X-ray spectroscope (EDS) was used to study the surface morphology and elementary composition. Transmission electron microscopy (TEM) was performed with a JEOL JEM 1010 to study the size and shape of powder samples. Photoluminescence (PL) spectra measurements were performed at room temperature by the Hitachi F-7000 fluorescence spectrophotometer using xenon lamp as an excitation source and the lifetime measurements were taken at the chopping speed of 20 Hz.
3 Results and discussion
3.1 XRD analysis
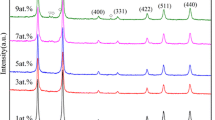
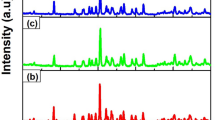
The XRD patterns of the prepared samples are shown in Fig. 1. The prepared samples are polycrystalline with a structure that matches the standard pattern of cubic ZnAl2O4 spinel (JCPDS 82-1043). This crystalline structure was expected to alter in the presence of RE ions due to the comparably larger size of the dopants at substitutional sites, but due to the low concentration of the dopants the structure was found to remain the same. Additional diffraction peaks were not observed, suggesting no Eu3+- and/or Gd3+-related impurities and indicating a successful substitution of either Zn2+ or Al3+ by the Eu3+ and/or Gd3+ ions within the host lattice. Thus, the presence of symmetric and strong diffraction peaks indicates the growth of monophase ZnAl2O4 crystallites at an annealing temperature of 800 ºC.

The XRD patterns for the a host, singly doped (with 0.1% Eu3+ and 0.1% Gd3+) and b ZnAl2O4:0.1% Eu3+; x% Gd3+ (0 ≤ x ≤ 1.2) co-doped samples
The analysis of the 311 diffraction peaks for the prepared samples is presented in Fig. 2. It is noted that the incorporation of foreign atoms into the host matrix influences the diffraction intensity and peak angle. The decrease or increase of the diffraction intensity in Fig. 2 can be attributed to the loss or gain of crystalline quality [12]. When comparing the peak diffraction angle of the host and singly doped samples in Fig. 2a, there is a slight peak shift to higher diffraction angle, which indicates a slight decrease in the lattice parameters. The ionic radius of both Zn2+ (0.74 Å) and Al3+ (0.53 Å) [7] is smaller than that of Eu3+ (0.95 Å) [13] and Gd3+ (1.08 Å) [3]. It is predicted by Vegard’s law [14, 15] that if the incorporation process takes place via substitution, the addition of Eu3+ and/or Gd3+ should expand the lattice. Figure 2a shows a clear indication of the violation of Vegard’s law. Similar results have been previous observed in the ZnAl2O4:Pb2+ system [16], where at lower Pb2+ concentration the lattice parameter decreased. This behaviour was attributed to the shrinkage of the Pb2+ outer electron shell due to its electronic interactions with neighbouring Zn2+ ions. Figure 2b shows the variation in diffraction angle of the co-doped sample when varying the concentration of Gd3+ ion (0 ≤ x ≤ 1.2). The peaks seem to shift to a lower diffraction angle indicating an increase in the lattice angle, expect for x = 0.1%, and the reason for this behaviour is currently unknown. The results displayed in Fig. 2b are in agreement with Verard’s law. The lattice parameters increase as predicted by Vegard’s law. The lattice parameters calculated from the most intense peak (311) are presented in Table 1 and correspond to the lattice parameter of ZnAl2O4 reported by Motloung et al. [10].

Analysis of the diffraction peak corresponding to the (311) plane in the a host, singly doped and b dual-doped samples
The crystallite sizes of the prepared nanocrystals were calculated using Scherrer's equation [17] and are presented in Table 1. Singly doping and co-doping slightly increased the crystallite sizes. Generally, and as expected, it is also noted that the higher (311) diffraction intensities (from Fig. 2) corresponded to bigger crystallite sizes.
3.2 Energy-dispersive X-ray spectroscopy
EDS was used to confirm the chemical composition of the host and co-doped (x = 1.2%) samples as shown in Fig. 3a, b, respectively. For the host samples shown in Fig. 3a, the expected elements, namely Zn, Al and O, are observed. At concentration x = 1.2% for the co-doped sample shown in Fig. 3b, Gd was also observed. The element Eu was not detected in Fig. 3b, which might be due to the low concentration of Eu3+ ions used in this study. In all EDS spectra, the additional peak of carbon (C) is due to the sample coating. No additional impurity peaks were detected, which agrees very well with the XRD results in Fig. 1.

The EDS spectrum of the a ZnAl2O4 (host) and b x = 1.2% nanophosphors
Figure 4 shows the EDS mapping for the x = 1.2% sample. The images of individual elements are displayed around the layered image, which suggest that the individual elements are homogeneously distributed on the surface.

EDXS elemental maps of x = 1.2% co-doped sample
3.3 Scanning electron microscopic analysis
SEM micrographs of the host, singly and co-doped samples are shown in Fig. 5. Figure 5a of the host reveals that the morphology consists of a rough surface with irregular particles of different sizes distributed all over the surface. Figure 5b of the 0.1% Eu3+-doped sample shows a similar morphology, with a smaller number of irregular particles. Figure 5c shows the 0.1% Gd3+-doped sample which has an intense agglomeration of particle over the surface. Figure 5d depicts the morphology of the x = 1.2% co-doped sample, which shows clusters of particles of different sizes randomly distributed. This morphology resembles both the morphologies presented in Fig. 5b, c.

The SEM micrographs of a host b singly doped with 0.1% Eu3+ c 0.1% Gd3+ and d co-doped x = 1.2% nanophoshor
3.4 High-resolution transmission electron microscopic analysis
The host, 0.1% Eu3+, 0.1% Gd3+ and x = 1.2% samples were examined by HR-TEM to confirm the crystallite size and shape. Their respective images are presented in Fig. 6. It can be seen that the crystallites are agglomerated to each other and their average crystallite size is around 20 nm, which confirms the values predicated by the XRD as displayed in Table 1.

HR-TEM images for the a host, b 0.1% Eu3+, c 0.1% Gd3+ and d x = 0.6% samples
3.5 Photoluminescence analysis
To determine the appropriate excitation wavelength, the host sample was excited with different excitation wavelengths ranging from 260 to 310 nm. The emission spectra illustrated in Fig. 7a show that the emission peaks are located around 393 nm. The peaks at around 550, 565, 593, 605 and 631 nm are due to the second-order (2nd order) emissions corresponding to 270, 280, 290, 300 and 310, respectively. The 393 nm emission originates within the intrinsic intra-band gap defects such as oxygen vacancies (\({V}_{o}^{*}\)) [13, 18]. The emission intensity as a function of excitation wavelength is shown in Fig. 7b. The Gaussian fit suggests that the optimum excitation wavelength is 284 nm.

a The emission spectra of the ZnAl2O4 (host) excited at various excitation wavelengths and b emission intensity as a function of excitation wavelength
The excitation and emission spectra of the host and singly doped samples are shown in Fig. 8a. The excitation spectra were taken when monitoring the violet emission at 393 nm. The excitation spectrum consists of only one band located at 284 nm (corresponds to the optimum excitation shown in Fig. 7b), which can be attributed to the band-to-band transition of AlO6 anion grouping in ZnAl2O4 [13, 19]. Note that the emission spectra were taken when monitoring the 284 nm excitation wavelength. The emission spectrum consists of four emission peaks located at 393, 400, 578 and 618 nm. The emission at 393 and 400 nm can be attributed to the intrinsic intra-band gap since the emission at around 393–400 nm (violet) is not the characteristic of the Gd3+ spectroscopy [20]. The emission peak at 578 nm is a combination of the 5D0 → 7F1 Eu3+ transition [14, 16] and the second order (2nd order) corresponding to the 284 nm excitation wavelength. The peak at 618 nm can be attributed to the 5D0 → 7F2 transition within Eu3+ [14, 16]. To investigate the origin of each of the emission bands observed in detail, the normalized emission spectra are shown in Fig. 8b. There is no peak shift observed when the host is singly doped with 0.1% Eu3+, which implies that the emission at 393 nm is from the host. However, when the host is singly doped with 0.1% Gd3+, it is clear that there is a slight peak shift from 393 to 400 nm, which signifies that the trap levels responsible for the emission at 393 nm has slightly transcended downwards within the Eg [20, 21]. Figure 8c depicts the emission and excitation of the host and co-doped sample. Similar excitation and emission peaks to that of Fig. 8a are observed. Figure 8d presents the violet emission intensity at 393 nm as a function of Gd3+ concentration, which has been fitted by a second exponential decay behaviour. Figure 8e shows the zoomed emission region near 618 nm, and the results show that for fixed Eu3+ concentration (0.1%), the x = 0.4% is an optimum Gd co-dopant concentration for the Eu3+ 5D0 → 7F2 emission. Since the Gd3+ ions would not be excited at 393 nm, this effect is attributed to physical changes in the nanocrystalline phosphor as a result of Gd co-doping.

Excitation and emission spectra of the a host and singly doped samples, b normalized emission for the host and singly doped samples, c co-doped samples at varying Gd3+ concentrations (0 ≤ x ≤ 1.2), d emission intensity of the peak located at 393 nm as a function of Gd3+ concentration and e the zoomed emission of peaks located at 618 nm
The x = 0.4% co-doped sample was further excited at various wavelengths in the range of 260 to 310 nm and the emission spectra are shown in Fig. 9a. In addition to the host defect emission peak at 400 nm and Eu3+ peak at 618 nm discussed above, some additional second-order peaks which shifted, corrosponding to double the excitation wavelengths, were observed. The emission intensity at 618 nm as a function of excitation wavelength is depicted in Fig. 8b. A Gaussian fit shows that to optimize the emission from Eu3+, the corresponding excitation must be 263 nm. The excitation of Eu3+ at 263 nm is attributed to the charge transfer transition (CT) which is an electron transfer from the O2− (2p6) orbital from the host to the Eu3+ empty 4f6 orbital [22, 23].

a The emission spectra of the x = 0.4% sample excited at various excitation wavelengths and b emission intensity as a function of excitation wavelength
The luminescence mechanisms for all emissions arising from the prepared samples are illustrated in Fig. 10. The location of the Eu3+ ion transitions within the host band gap was placed according to Dorenbo’s diagram [24, 25]. The absence of the energy transfer from Eu3+ → Gd3+ and charge CT from the host → Gd3+ serves as the main reasons why there is no emission from Gd3+ at all. In addition and based on the Dorenbo’s diagram [24,25,26,27], the results also suggest that the Gd3+ is located where there are no probabilities of trapping the de-excited electron within the host material. As a result, the Gd3+ energy levels are not shown in the emission mechanism.

The proposed excitation and emission pathway mechanism in ZnAl2O4:x% Eu3+
3.6 Lifetime study
The radiative lifetimes of the defect emission from the prepared samples at 393 nm were measured using excitation at 284 nm. The lifetimes were fitted with the first-order exponential decay [3]:
where \(I\) represents the emission intensity, \(A\) is the background, \(B\) is a fitting parameter and τ is the decay time. The results are shown in Fig. 11 and the values are presented in Table 1. The lifetime of ~ 515 ms is orders of magnitude longer than that characteristic for RE emissions and indicates that carrier trapping occurs at defects in the host. Since the emission occurs on a timescale which is short compared to phosphors which are used commercially for persistent luminescence (hours), the traps are relatively shallow and detrapping occurs comparatively quickly at room temperature.

Decay curves on logarithmic intensity scale of the a host and singly doped samples, and b co-doped samples
3.7 Colour chromaticity
The International Commission on Illumination (CIE) colour chromaticity coordinates [28] of the prepared powders are illustrated in Fig. 12. Figure 12a illustrates the chromaticity co-ordinates of the host sample excited at different wavelengths, which falls in the violet region as expected and is not significantly changed by varying the excitation wavelength. Figure 12b illustrates the chromaticity co-ordinates of the host and singly doped (with 0.1% Eu3+ and 0.1% Gd3+) excited at 284 nm. Only small shifts occur, which correspond to a slight change in the peak emission wavelength of the defect emission to ~ 400 nm when Gd is added as dopant, or a weak red emission at 618 nm when Eu is added as dopant. This weak influence might be due to the low concentration of dopants used for the singly doped samples. However, Koao et al. [29] showed that an increase in Eu3+ concentration on the ZnO system shifts the emission colour towards the red emission colour. Figure 12c illustrates that the chromaticity co-ordinates for the co-doped nanophosphors and their colour coodinates are presented in Table 1. Only a small effect on the CIE colour chromaticity occurs when varying the Gd3+ concentration. Figure 12d shows that the emission colour from the x = 0.4% nanopowder sample shifts a little when excited at different wavelengths and moves slightly towards the centre when excited near 260–270 nm, corresponding to when the red emission from Eu3+ is the strongest (Fig. 9b).

CIE colour diagrams for a the host at varying wavelengths, b the host and singly doped samples excited at 284 nm, c co-doped samples excited at 284 nm and d x = 0.4% co-doped samples at varying excitation wavelengths
4 Conclusions
A series of ZnAl2O4 nanophosphors was successfully prepared by using a citrate sol–gel technique. The undoped host and samples singly doped with either Eu or Gd (0.1%), as well as co-doped samples with 0.1%Eu3+ and x% Gd3+ (0 ≤ x ≤ 1.2) were studied. The XRD and HR-TEM analysis confirmed that all of the nanopowders consisted of the single-phase cubic spinel structure. EDS was able to confirm the elemental compositions of the prepared sample. SEM shows that both singly doping and co-doping influenced the host morphology. PL spectra showed an emission peak at ~ 393–400 nm, which originated from the host defects and was slightly shifted as a result of Gd doping, as well as an emission at 618 nm when Eu3+ ions were present. The CIE colour chromaticity showed that although slight colour shifts occur with doping and excitation wavelength, the colour in the violet region is dominated by the strong host defect emission, which has a long lifetime associated with trapping centres.
References
R. Roesky, J. Weiguny, H. Bestgen, U. Dingerdissen, App. Catal. A 176, 213–220 (1999)
M. Zawadzki, Sol. State Sci. 8(1), 14–18 (2006)
V. Singh, R.P.S. Chakradhar, J.L. Rao, D.K. Kim, J. Lumin. 128(3), 394–402 (2008)
J. Popovic, B. Grzeta, B. Rakvin, E. Tkalcec, M. Vrankic, S. Kuranjica, J. Alloys Comp. 509(34), 8487–8492 (2011)
G. Lakshminarayana, L. Wondraczek, J. Sol. State Chem. 184(8), 1931–1938 (2011)
W.M. Mulwa, B.F. Dejene, M.O. Onani, C.N.M. Ouma, J. Lumin. 184, 7–16 (2017)
W. Strek, P. Deren, A. Bednarkiewicz, M. Zawadzki, J. Wrzyszcz, J. Alloys Comp. 300–301, 456–458 (2000)
D.A. Zatsepin, D.W. Boukhvalov, A.F. Zatsepin, YuA Kuznetsova, M.A. Mashkovtsev, V.N. Rychkov, V.Y. Shur, A.A. Esin, E.Z. Kurmaeva, App. Sur. Sci. 436, 697–707 (2018)
R.K. Tamrakar, K. Upadhyay, Optik 143, 125–130 (2017)
S.V. Motloung, F.B. Dejene, R.E. Kroon, H.C. Swart, O.M. Ntwaeaborwa, Phys B 468–469, 11–20 (2015)
T. Chengaiah, C.K. Jayasankar, L.R. Moorthy, Phys B 431, 137–141 (2013)
Q. Hou, F. Meng, J. Sun, Nano. Res. Lett. 8, 144 (2013)
B. Cheng, S. Qu, H. Zhou, Z. Wang, Nanotechnology 17, 2982–2987 (2006)
C. -Perez, J. Lambert, A. Alatorre-Ordaz, J.A. Gutierrez, T. Lopez-Luke, R. Ramirez-Fuentes, T. Kobayashi, J. Lumin. 184, 123–129 (2017)
A. Nakrela, N. Benramdane, A. Bouzidi, Z. Kebbab, M. Medles, C. Mathieu, Res. Phy. 6, 133–138 (2016)
S.V. Motloung, F.B. Dejene, H.C. Swart, O.M. Ntwaeaborwa, J. Sol-Gel Sci. Technol. 70(3), 422–427 (2014)
V.M. Maphiri, F.B. Dejene, S.V. Motloung, Res. Phy. 7, 3510–3521 (2017)
B.S. Barros, P.S. Melo, R.H.G.A. Kiminami, A.C.F.M. Costa, G.F. de Sá, S. Alves, J. Mater. Sci. 41(15), 4744–4748 (2006)
H. Dixit, N. Tandon, S. Cottenier, R. Saniz, D. Lamoen, B. Partoens, V. Van Speybroeck, M. Waroquier, New J. Phy. 13, 063002 (2011)
Q. Wang, S.-Y. Ouyang, W.-H. Zhang, B. Yang, Y.-P. Zhang, H.-P. Xia, Acta Metall. Sin (Engl. Lett.). 28, 487491 (2015)
P. Gupta, A.K. Bedyal, V. Kumar, Y. Khajuria, S.P. Lochab, S.S. Pitale, O.M. Ntwaeaborwa, H.C. Swart, Mat. Res. Bull. 60, 401–411 (2014)
W. Jiao, Z. Zhijun, Z. Jingtai, J. Rare Earths 33(12), 1241–1245 (2015)
M.K. Lau, J. Hao, Energy Procedia 15, 129–134 (2012)
I.A.M. Ibrahim, Z. Lences, P. Sajgalik, L. Benco, J. Lumin. 164, 131–137 (2015)
Y. Shimizu, K. Ueda, J. Lumin. 168, 14–19 (2015)
J.I. Eldridge, J. Lumin. 214, 116535 (2019)
S.V. Motloung, K.G. Tshabalala, R.E. Kroon, T.T. Hlatshwayo, M. Mlambo, S. Mpelane, J. Mol. Struct. 1175, 241–252 (2019)
hhttps://www.mathworks.com/matlabcentral/fileexchange/29620-cie-coordinate-calculatori 2012.
L.F. Koao, B.F. Dejene, H.C. Swart, S.V. Motloung, T.E. Motaung, Opt. Mater. 60, 294–304 (2016)
Acknowledgements
This work was supported by the South African National Research Foundation (NRF) Thuthuka programme (Fund number: UID99266) and NRF incentive funding for rated researchers (IPRR) (Grant no: 114924). The author would also like to acknowledge Mr. T.M Manamela for the sample synthesis and Dr James Wesley-Smith at Electron Microscopy Unit at Sefako Makgatho Health Science University for the SEM and TEM imaging.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Maphiri, V.M., Dwivedi, Y., Koao, L.F. et al. Analysis of varying Gd3+ concentrations on the structure and optical properties of ZnAl2O4:0.1% Eu3+; x% Gd3+ (0 ≤ x ≤ 1.2) synthesized via citrate sol–gel method. Appl. Phys. A 126, 73 (2020). https://doi.org/10.1007/s00339-019-3260-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-019-3260-y