
Abstract
Electronic and nonlinear optical properties of transition metal (Sc, Fe, Cu, and Zn) substitutional doped C20 fullerenes are studied through DFT calculations. Replacement of carbon of C20 fullerene with a transition metal atom remarkably increases the hyperpolarizability (βo) of the system, compared to 0 au hyperpolarizability of pure C20 fullerene. The maximum βo is calculated for Sc–C19 (2224.5 au) followed by Fe–C19 (790.1 au), Cu–C19 (592.1 au), and Zn–C19 (564.6 au). The same order is found for the polarizability of the resulting systems. The polarizability and hyperpolarizabilities are found to decrease with increase in ionization potential of the doped transition metal. Molecular reactivity descriptors reveal that the iron-doped fullerene Fe–C19 is the softest system with the highest electrophilicity among all studied. The outcome of this study will be useful for promoting the possible use of the metal–fullerene systems as a new type of electronic nano-devices having good-performance nonlinear optical (NLO) properties.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
During the last few decades, the design of new materials with outstanding NLO properties has received much attention due to their potential applications in optical and electro-optical devices [1,2,3,4,5,6,7,8]. A number of strategies have been reported in the literature to enhance the nonlinear optical properties of organic and inorganic materials. Electron push–pull mechanism (for organic NLO materials), metal organic assemblies and formation of electrides are few strategies to mention in this regard. Recently, it has also been found that the introduction of diffuse excess electrons (through doping with alkali metal atoms) in a system is an effective strategy for enhancement of NLO properties [9,10,11,12,13]. Systems with diffuse excess electrons have significantly large hyperpolarizability (βo) values. Alkali metal atoms are excellent at introducing diffuse excess electrons in a system. Excess electrons from metal possibly lower down the excitation energy and, therefore, enhance the NLO response [10, 13,14,15]. Doping of a structure with certain atoms is a way of enhancing the electronic property of that structure [16,17,18,19,20,21].
The literature reveals a number of examples where electride or diffuse excess electron approach has been applied to obtain exceptionally large hyperpolarizability values [6,7,8]. For example, Niu et al. [6] obtained stable inorganic electride compounds by doping three alkali metal atoms (Li, Na, and K) on the exterior surface of fullerene-like Al12N12 nanocage. They found large first hyperpolarizability values for the studied metal cluster. In another study, Ayub and coworkers [7] demonstrated how the substitutional doping of the alkali metal atom affects the hyperpolarizability of BN nanocages. They found improvement in the NLO properties and first hyperpolarizability of the resulting clusters upon metal incorporation. Substitutional doping of alkali metal in AlN nanocages has also been reported by the same authors [8]. The hyperpolarizabilities of the doped systems range from 103 to 105 au, compared to 0 au for parent undoped systems. For the last few years, modified carbon structures have gained much attention due to their unique electronic properties [22,23,24].
Among carbon nanostructure, the C20 is the smallest fullerene with a dodecahedral cage structure. C20 has gained substantial attention for many physical applications due to its high surface area. Prinzbach et al. [25] synthesized the C20 fullerenes for the first time. The C20H20 was converted to C20Br20 by substituting the H atoms with Br atoms and then de-brominated to synthesize gas-phase C20 fullerene. The C20 was synthesized in the solid phase by using ion beam irradiation [26] and laser ablation [27] methods. It has been revealed that functionalization and doping can modify the electronic and magnetic properties of the C20 and related nanostructures [28,29,30]. An et al. [28] found that the unstable C20 cage can be stabilized by doping the alkali metal atoms (Li, Na, K, Rb, and Cs) at the center of the nanocage (encapsulation). They theoretically investigated (through DFT the transport properties of the C20 and the endohedral Li@C20 metallofullerene coupled to three-dimensional Au electrodes [29]. In another study, using DFT calculations, the impacts of transition metal atoms (Sc, Ti, V, Cr, Mn, Fe, Co, Ni, Cu, and Zn) doping at the center of C20 fullerene were explored [30]. The results reveal that the doping of Co, Ni, Cu, and Zn atoms is endothermic. On the other hand, the doping of Sc, Ti, V, Cr, Mn, and Fe atoms is exothermic. Recently, Rad et al. [31] used DFT calculations to study the application of transition metal-substituted C20 for the adsorption of adenine thymine and uracil [32] nucleotides. They found that transition metals are the best choice for increasing the electronic property of C20 fullerene. In the research conducted by Dheivamalar and Sugi [33], the structures and stabilities of dodecahedral fullerene C19X (X = Ni, Ti) and C20 were investigated using DFT. Paul et al. [34] studied the stabilities and reactivities of Cu- and Zn-doped structures of fullerene C20 by the reactivity parameters such as chemical hardness, chemical potential, and electrophilicity index. They found the maximum hardness principle and minimum electrophilicity principle for those metal–fullerenes. The same group [35] compared the global reactivity of C19X (X = Fe, Co) fullerenes with C20 fullerene using the DFT approach. They confirmed that the applicability of fullerenes significantly increases by transition metal doping.
The literature is quite rich in examples where electronic, spectroscopic and nonlinear optical properties of a system are significantly influenced by doping [6,7,8,9,10,11,12,13,14,15]. This has been reported for carbon fullerenes, inorganic fullerenes, organometallic compounds, small clusters, etc. C60 fullerene and higher analogs have been extensively studied for doping’ however, the smaller C20 fullerene has not been well explored. Fullerenes can be doped with an external atom/molecule in three different ways: exohedral, endohedral and substitutional. In exohedral and endohedral doping, the external atom interacts non-covalently. On the other hand, the external atom has a covalent bond in substitutional doping because it involves the replacement of an atom of fullerene with a dopant (transition metal in our case). Substitutional doping is relatively less studied compared to exohedral and endohedral doping. So, in this study we were motivated to further follow the substitution doping of fullerene. Substitution doping involves the replacement of one C atom of C20 with a dopant (transition metal atom).
It should be noted that transition metal doping on fullerene provides various functional properties including the ability of light absorption, catalytic activity, and giving and removing electron capability. These features would absolutely influence and moderate the useful characteristics of the resultant doped fullerene. Solar-cell materials could also be considered through the transition metal-doped fullerenes [34].
In this regard, previously our group had investigated the NLO property of substitutional doping of the second row transition metal including zirconium, molybdenum, ruthenium, and palladium as an operative technique to advance the NLO and electronic property of C20 fullerene [36]. In another research, we studied the NLO and the electronic properties of first row transition metals including Cr-, Ni-, and Ti-substituted C20 fullerenes [37]. We noticed that each transition metal in this row has its own effect on the NLO as well as the electronic property of C20 fullerene. Therefore, we were motivated in this study to see the effect of the other first row transition metals (Sc, Fe, Cu, and Zn) on the NLO properties of C20.
C20 fullerene is a symmetric fullerene for which hyperpolarizability is zero. C20 is an interesting molecule because this is the smallest fullerene which breaks the “isolated pentagon rule”. Replacement of a carbon atom of fullerene with a dopant, more particularly, the transition metal, is expected to break the symmetry due to the elongated M–C bond. Moreover, this doping is expected to produce improved electronic and nonlinear optical properties. For this purpose, Sc, Fe, Cu, and Zn are doped into C20 fullerene. The selected dopants include examples from early, middle, and late transition metals.
The transition metal atoms chosen are representative of early, middle, and late transition metals. Another objective of the study is to explore the effect of doped transition atom on the geometry and charge allocation of the metallofullerene system. The outcomes of this study can be useful for designing new materials for their possible utilization in electronics and high-performance NLO materials.
2 Computational details
To study the electronic structure of metal–fullerene, one atom of C20 is substituted with transition metal atoms (Sc, Fe, Cu, and Zn) and the systems were allowed to fully relax (see Fig. 1). The optimization was achieved at meta-hybrid functional (ωb97XD) of DFT with 6–31G(d, p) basis set, as implemented in G09 [38]. ωB97XD is a long range and dispersion-corrected method to reliably estimate the electronic properties of related nanostructures [39]. The structures are allowed to fully relax until the force on each atom is less than 0.01 eV/Å. The true minima nature of these complexes was confirmed by the absence of any imaginary frequency. Different spin states were considered (four lowest possible) for different transition metal-doped fullerenes. The most stable spin states for transition metal-doped fullerenes are singlet for Zn–C19, doublet for Sc–C19 and Cu–C19, and quintet for Fe–C19.

Relaxed structure of each metallofullerene: Sc–C19, Cu–C19, Fe–C19, and Zn–C19
For the valuation of stability of pure and doped C20, cohesive energies (Eco) are calculated by the following equations:
Cohesive energies (Eco) [7]:
where N is the number of atoms in the system and EC20, EC, EM, and EC19M are the energy of C20 fullerene, single carbon atom, metal, and metallofullerene, respectively.
Par et al. [40] stated the concept of electrophilicity in 1999. The chemical potential (µ) is defined based on the following equation [41]:
Here, EHOMO is the energy of the highest occupied molecular orbital (HOMO) and ELUMO is the energy of the lowest unoccupied molecular orbital (LUMO). Additionally, hardness (ŋ) can be described using the Koopmans’ theorem [41] as:
Softness (S) [41] and electrophilicity (ω) [41] are also stated as in the following equations, respectively:
Polarizability calculations were carried out at the same level of theory [ωB97XD/6–31G(d, p)]. However, the first hyperpolarizability was calculated at ωB97XD/6–311 + G(d,p). The mean polarizability (α) and the first hyperpolarizability (βo) are denoted as [7]:
3 Results and discussion
3.1 Geometrical parameters
The optimized geometries are analyzed to realize the effect of dopant on the structural parameters. First of all, the most stable spin states of the doped fullerenes are evaluated. For this purpose, the four lowest possible spin states (such as doublet, quartet, sextet, and octet) of all doped fullerenes are considered and the relative energies are compared. All doped fullerenes are stable in the lowest spin state except Fe–C19, for which quintet spin state is the most stable. Different spin states of these fullerenes do not allow for a direct comparison of the bond lengths. More precisely, no trend could be traced in the M–C bond lengths.
Analysis of the optimized geometries reveals that the symmetry of the fullerenes is completely broken. The transition metal element is also not present at the center. The M–C bond lengths vary among themselves in a system. The Zn–C19 is the system with the least variation of bond lengths. The Zn–C bond lengths are 1.989 and 1.990 Å. The Sc–C bond lengths are 2.114 and 2.148Å, whereas the corresponding bond lengths in Cu–C19 are 1.931 and 1.960Å. The Fe–C bond lengths are 1.989, 1.998, and 2.001Å. In general, the M–C bond lengths decrease with increase in atomic number except for Zn–C19. This behavior is attributed to the atomic radii of the transition metals concerned. The atomic radius of the transition metal atoms decreases from Sc to Cu and then increases for Zn. For example, the atomic radii of Sc, Fe, Cu, and Zn are 1.44, 1.16, 1.16 and 1.24 Å, respectively. The longer bond length of Zn–C19 is also reflective of the weak interaction between fullerene and the metal atom (vide infra). A similar trend is observed in the diameter of the doped fullerenes. The diameter of the fullerenes is measured from the metal atom (as shown in Fig. 1). The diameter of the fullerene decreases while moving in the first row of the transition metals, except for Zn. The diameters of Sc–C19, Fe–C19, Cu–C19, and Zn–C19 are 5.142, 5.039, 4.934 and 4.971Å, respectively.
Cohesive energy is next analyzed to account for the trends in bond lengths (see Table 1). The cohesive energy of C20 fullerene is 8.01 eV which decreases for all doped systems. The cohesive energy of Sc–C19 is 7.78 eV. The cohesive energy decreases while moving along the periodic table except for Cu–C19 where a slight increase in the cohesive energy is observed. The cohesive energies of Fe–C19, Cu–C19, and Zn–C19 are 7.69, 7.71 and 7.59 eV, respectively. The cohesive energy of Zn–C19 is much lower than that of the other doped systems, which reflects its relative instability. This observation is consistent with the trends of bond lengths where the higher M–C bond reflected the instability of the Zn–C19 fullerene. The results also illustrate that the decrease in M–C bond length along the period (from Sc to Fe to Cu) is merely due to the small size of atoms along the series. It may be argued that the cohesive energies are intrinsically dependent on the strength of the M–C bond. The M–C bond strength is the lowest for Zn (about 22 kcal mol−1) compared to 28–33 kcal mol−1 for M–C (M = Sc, Fe, and Cu). This low bond strength might be responsible for low cohesive energies.
3.2 Charge analysis
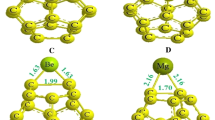
NPA charges are also analyzed for better understanding of the bonding between transition metal and fullerene. The NPA charges on the transition metal decrease from Sc to Cu, but then again increase for Zn. The NPA charges on the transition metals are 1.634, 1.350, 1.213, and 1.581 e for Sc–C19, Fe–C19, Cu–C19 and Zn–C19, respectively which are much higher than that reported for Ni–C19 (0.971 e) and Cr–C19 (1.044 e) [31, 32]. The fullerene parts in these structures possess a negative charge. The strength of the negative charge on carbon is proportional to the intensity of the positive charge on the transition metal. The highest charge on the carbons (neighboring transition metal) is observed for Sc–C19 (− 0.409 and − 0.506). It is interesting to note that all carbon atoms in the vicinity of the transition metal do not possess the same charge, which reflects the unsymmetrical nature of these doped fullerenes. Similar unsymmetrical distribution of charges is also observed for Fe–C19 and Cu–C19. The Zn–C19 fullerene is the only system where identical charges are seen on all carbon atoms, in the vicinity of the transition metal (See Fig. 2). It is also worth mentioning that almost all carbon atoms of the fullerene moiety in these doped structures possess negative charges; however, the intensity of the charge decreases while moving away from the transition metal.

NPA charge distributions of different metal–C20 clusters: Sc–C19 (a), Cu–C19 (b), Fe–C19 (c), and Zn–C19 (d)
3.3 Electronic properties
The electronic properties of transition metal-doped C20 fullerenes are next calculated (see Table 1). Figure 3 shows the HOMO and LUMO distributions of all studied systems. As can be seen in Fig. 3, substitution doping of transition metal on C20 fullerene results in significant changes in both HOMO and LUMO distributions. C20 fullerene has a relatively large HOMO–LUMO gap (5.441 eV). The energies of HOMO and LUMO are − 7.33 and − 1.889 eV, respectively. The doping of transition metal affects the HOMO–LUMO gap; however, the behavior of early and late transition metals is quite different. Early transition metal-doped C20 fullerenes have a HOMO–LUMO gap lower than the bare C20 fullerene. HOMO–LUMO gap of Sc–C19 is 5.241 compared to 5.441 eV for C20. The energies of HOMO and LUMO for Sc–C19 are − 6.436 and − 1.195 eV, respectively, compared to − 7.330 and − 1.889 eV for C20. Analysis of the results reveals that the doping of Sc increases the energies of HOMO and LUMO; however, the effect is more pronounced on the former which leads to a decrease in the HOMO–LUMO gap. The distribution of densities also reveals that the HOMO and LUMO are both spread over the entire skeleton including the Sc atom. The distribution of density in HOMO on transition metal suggests a strong overlap between the metal center and the carbon part. Another supporting evidence for strong interaction between the metal and carbon part is the increase in the energy of HOMO. These findings are consistent with charge analysis and thermodynamic analysis which suggest that the carbon fragment and Sc atom interact strongly. Doping of an iron atom has a quite intense effect on the energies of HOMO and LUMO (different from the doping of other atoms). The energy of HOMO is increased, whereas the energy of LUMO is decreased which causes the HOMO–LUMO gap to decrease to 2.737 eV. The energies of HOMO and LUMO are − 5.708 and − 2.971 eV, respectively. Doping with a late transition metal (Cu or Zn) causes the HOMO–LUMO gap to increase. The increase in the HOMO–LUMO gap is due to a decrease in the energy of HOMO and an increase in the energy of LUMO, although the former is more pronounced. Moreover, the effect of Zn is greater than the effect of Cu (see Fig. 3). The HOMO–LUMO gaps for Cu–C19 and Zn–C19 are 6.165 and 6.365 eV. The energies of HOMO and LUMO for Cu–C19 are 7.878 and − 1.713 eV, whereas the corresponding values for Zn–C19 are − 7.994 and − 1.629 eV, respectively. Zn–C19 is unlike the other three nanocages. For Zn–C19, the zinc atom is deprived of any density in HOMO and LUMO, whereas in the other systems the HOMOs and/or LUMOs have density spread over the metal atom. Therefore, it may be argued that the overlap between the Zn and carbon framework is not strong which leads to low cohesive energies.

HOMO–LUMO distributions of different systems
3.4 Nonlinear optical properties
Nonlinear optical properties of these doped fullerenes are evaluated through the first and second hyperpolarizability values. Besides the first hyperpolarizability, polarizability values are also calculated and shown in Table 2. C20 fullerene is a spherical molecule with relatively small polarizability. The polarizability of C20 is 186.1 au, which increases when doped with transition metals. The highest polarizability is calculated for Sc–C19 (222.1 au) which decreases to 209.9 au for Fe–C19. The high polarizability of Sc–C19 over Fe–C19 may be attributed to a relatively large M–C bond or the large size of the Sc atom. The polarizability for Cu–C19 and Zn–C19 are 205.6 and 204.81 au. The polarizability of Zn–C19 is lower than that of Cu–C19 which is contrary to the expected based on the M–C bond length. Therefore, the size of the transition metal atom is a more probable reason for the trend. The polarizability values decrease monotonically with an increase in the atomic number of the doped atom (along the row).
The hyperpolarizability also follows a similar monotonic trend where the hyperpolarizability values decrease while moving along the row of the periodic table. The highest hyperpolarizability of 2224.5 au is calculated for Sc–C19, followed by 790.1 au for Fe–C19. The hyperpolarizability of Cu–C19 and Zn–C19 is 592.1 and 572.0 au, respectively. The hyperpolarizability of alkali metal-doped fullerenes is well known in the literature [7, 8, 10, 11]. The remarkably high hyperpolarizability of alkali metal-doped fullerenes is believed to arise from diffuse excess electrons arising from the interaction of alkali metal with the rest of the cluster. The generation of the diffuse excess electron depends on the ionization potential of the doped metal. The lower ionization potential of doped metal atom favors the generation of diffuse excess electrons.
Quite similar to the alkali metal-doped cases [7, 8], it may be argued here that the decrease in hyperpolarizability along the row of first transition metal atoms is due to increase in the ionization potential. Scandium, due to its large size, loses electrons and causes an increase in hyperpolarizability. The zinc atom, on the other hand, is very resistant to loose electron and, therefore, is not a stimulant for boosting hyperpolarizability.
3.5 Quantum molecular descriptors
The global indices of reactivity are next studied according the method presented by Rad et al. [32] for all metallofullerenes to realize the reactivity and stabilities of the structures. The vertical ionization potential is obtained by considering the difference in energy of a neutral molecule and its cation (with the geometry of the ground state). Similarly, the energies of neutral and anionic species are taken for electron affinities. As can be seen in Table 3, the ionization potential of C20 is 7.39 eV which decreases to 6.40 and 7.31 eV for Sc–C19 and Fe–C19, but increases to 7.61 and 7.75 eV for Cu–C19 and Zn–C19. Quite similar to the ionization potential, electron affinities also do not follow any regular trend. The electron affinity of C20 fullerene is 1.91 eV which decreases to 1.26 eV for Sc–C19; however, it increases remarkably to 2.41 eV for Cu–C19.
The hardness of the doped structures follows the same trend as for their ionization potential. The hardness first decreases from C20 to Sc–C19 (and Fe–C19) and then increases for Cu–C19 and Zn–C19. The hardness of C20 is 2.72 eV, whereas the hardness for Sc–C19 and Fe–C19 is 2.62 and 1.368 eV, respectively. Zn–C19 is the hardest cluster among all with a hardness value of 3.182 eV. Since softness is the opposite of hardness, therefore, it was expected that the softness would follow an opposite trend to hardness. Indeed, this is the case. The Fe–C19 is the softest cluster with a softness value of 0.365 eV. The Zn–C19 is the least soft material in the series with softness values of 0.157 eV. The softness of Sc–C19 and Cu–C19 are 0.191 and 0.162 eV, respectively (see Table 3).
The chemical potential of C20 fullerene is − 4.609 eV which decreases to − 3.815 eV for Sc–C19. The chemical potential of Sc–C19 is lower in the series. The chemical potential of Fe–C19 is higher than that of Sc–C19, but lower than that of the bare C20 fullerene. The chemical potentials of late transition metal-doped fullerenes (Cu–C19 and Zn–C19) are higher than those of the parent fullerene. The chemical potentials of Cu–C19 and Zn–C19 are − 4.765 and − 4.811 eV. The electrophilicity, which is dependent on the chemical potential and hardness, is also calculated. The higher chemical potential and low hardness lead to high electrophilicities. The highest electrophilicity is calculated for Fe–C19 (6.872 eV). The high electrophilicity of Fe–C19 is due to the lower hardness (1.368 eV). The electrophilicity of other doped fullerenes is lower than that of the parent C20. The electrophilicity values of Sc–C19, Cu–C19, and Zn–C19 are 2.780, 3.725, and 3.634 eV compared to 3.909 eV for C20. It can be concluded from a quantum molecular descriptor that Fe–C19 is the most electrophilic in nature.
4 Conclusion
Substitution of a carbon atom of C20 fullerene with a transition metal atom (Sc, Fe, Cu, and Zn) toward following the change in electronic and NLO properties has been investigated using DFT approach. For this purpose, the most stable spin states of the doped fullerenes were appraised. It was found that the cohesive energy of C20 fullerene decreases for all doped systems while moving along the periodic table except for Cu–C19 where a slight increase was observed. Our results show that the NPA charges on the transition metal decrease from Sc to Cu, but then again increases for Zn. The fullerene parts in these structures possess negative charge, and its strength is proportional to the intensity of the positive charge on the transition metal. The polarizability of C20 rises when doped with transition metals. The maximum polarizability and hyperpolarizability are calculated for Sc–C19 which decreased monotonically with an increase in the atomic number of the doped atom. The decrease in hyperpolarizability along the row of first transition metal atoms is due to an increase in the ionization potential.
References
M. Shabbir, T. Minami, H. Fukui, K. Yoneda, R. Kishi, Y. Shigeta, M. Nakano, J. Phys. Chem. A 116, 1417 (2012)
M. Shabbir, S. Ito, M. Nakano, R. Kishi, K. Yoneda, Y. Kitagawa, M. Shkir, A. Irfan, A.R. Chaudhry, S. AlFaify, A. Kalam, Phys. Chem. Chem. Phys. 17, 5805 (2015)
M. Shabbir, K. Fukuda, T. Minami, R. Kishi, Y. Shigeta, M. Nakano, Chem. Eur. J. 19, 1677 (2013)
M. Shabbir, H.L. Xu, R.L. Zhong, Z.M. Su, A.G. Al-Sehemi, A. Irfan, J. Mater Chem. C 1(35), 5439 (2013)
M. Shabbir, H. Xu, Y. Liao, Y. Kan, Z. Su, J. Am. Chem. Soc. 131, 11833 (2009)
M. Niu, G. Yu, G. Yang, W. Chen, X. Zhao, X. Huang, Inorg. Chem. 53, 349 (2014)
J. Iqbal, K. Ayub, J. Alloys. Compd. 687, 976 (2016)
J. Iqbal, K. Ayub, RSC. Adv. 6, 94228 (2016)
W. Chen, Z.R. Li, D. Wu, R.Y. Li, C.C. Sun, J. Phys. Chem. B 109, 601 (2005)
W. Chen, Z.R. Li, D. Wu, Y. Li, R.Y. Li, C.C. Sun, J. Phys. Chem. A 109(12), 2920 (2005)
W. Chen, Z.R. Li, D. Wu, Y. Li, C.C. Sun, F.L. Gu, Y. Aoki, J. Am. Chem. Soc. 128, 1072 (2006)
H.L. Xu, Z.R. Li, D. Wu, B.Q. Wang, Y. Li, F.L. Gu, Y. Aoki, J. Am. Chem. Soc. 129, 2967 (2007)
F.F. Wang, Z.R. Li, D. Wu, B.Q. Wang, Y. Li, Z.J. Li, W. Chen, G.T. Yu, F.L. Gu, Y. Aoki, J. Phys. Chem. B 112, 1090 (2008)
W. Chen, Z.R. Li, D. Wu, F.L. Gu, X.Y. Hao, B.Q. Wang, R.J. Li, C.C. Sun, J. Chem. Phys. 121, 10489 (2004)
Z.J. Li, F.F. Wang, Z.R. Li, H.L. Xu, X.R. Huang, D. Wu, W. Chen, G.T. Yu, F.L. Gu, Y. Aoki, Phys. Chem. Chem. Phys. 11, 402 (2009)
A.S. Rad, S.A. Aghouzi, N. Motaghedi, S. Maleki, M. Peyravi, Mol. Simulat. 42, 1519 (2016)
A.S. Rad, K. Ayub, Comput. Theor. Chem. 1138, 39 (2018)
M. Mirmotahari, E. Sani, A.S. Rad, M.A. Khalilzadeh, J. Biomol. Struct. Dyn. (2019). https://doi.org/10.1080/07391102.2018.1546233
A.S. Rad, H. Pazoki, S. Mohseni, D. Zareyee, M. Peyravi, Mater. Chem. Phys. 182, 32 (2016)
A.S. Rad, J. Theor. Appl. Phys. 10, 307 (2016)
A.S. Rad, S.M. Aghaei, Curr. Appl. Phys. 18, 133 (2018)
A.S. Rad, J. Alloys. Compd. 682, 345 (2016)
S. Gholami, A.S. Rad, A. Heydarinasab, M. Ardjmand, J. Alloys. Compd. 686, 662 (2016)
A.S. Rad, Phys. E 83, 135 (2016)
H. Prinzbach, A. Weiler, P. Landenberger, F. Wahl, J. Wörth, L.T. Scott, M. Gelmont, D. Olevano, B.V. Issendorff, Nature. 407, 60 (2000)
Z. Wang, X. Ke, Z. Zhu, F. Zhu, M. Ruan, H. Chen, R. Huang, L. Zheng, Phys. Lett. A 280, 351 (2001)
Z. Iqbal, Y. Zhang, H. Grebel, S. Vijayalakshmi, A. Lahamer, G. Benedek, M. Bernasconi, J. Cariboni, I. Spagnolatti, R. Sharma, Eur. Phys. J. B 31, 509 (2003)
Y.P. An, C.L. Yang, M.S. Wang, X.G. Ma, D.H. Wang, J. Clust. Sci. 22, 31 (2011)
Y.P. An, C.L. Yang, M.S. Wang, X.G. Ma, D.H. Wang, Curr. Appl. Phys. 10, 260 (2010)
M.T. Baei, A. Soltani, P. Torabi, F. Hosseini, Monatsh. Chem. 145, 1401 (2014)
A.S. Rad, S.M. Aghaei, E. Aali, M. Peyravi, Diam. Relat. Mater. 77, 116 (2017)
A.S. Rad, S.M. Aghaei, E. Aali, M. Peyravi, M. Jahanshahi, Appl. Organomet. Chem. 32, 4070 (2018)
S. Dheivamalar, L. Sugi, Spectrochim. Acta. A 151, 687 (2015)
D. Paul, J. Deb, B. Bhattacharya, U. Sarkar, Int. J. Nanosci. 16, 1760026 (2017)
D. Paul, J. Deb, B. Bhattacharya, U. Sarkar, AIP Conf. Proc. 1832, 050107 (2017). https://doi.org/10.1063/1.4980340
A.S. Rad, K. Ayub, Comput. Theor. Chem. 1121, 68 (2017)
A.S. Rad, K. Ayub, Mater. Res. Bull. 97, 399 (2018)
G09 software, Revision D.01, M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng, J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery, Jr., J.E. Peralta, F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R. Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski, G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, Ö. Farkas, J.B. Foresman, J.V. Ortiz, J. Cioslowski, and D.J. Fox, C.T. Wallingford (2009)
J.D. Chai, M. Head-Gordon, Phys. Chem. Chem. Phys. 10, 6615 (2008)
R.G. Parr, L.V. Szentpaly, S. Liu, J. Am. Chem. Soc. 121, 1922 (1999)
T. Koopmans, Physica. 1, 104 (1933)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rad, A.S., Ayub, K. Change in the electronic and nonlinear optical properties of Fullerene through its incorporation with Sc-, Fe-, Cu-, and Zn transition metals. Appl. Phys. A 125, 430 (2019). https://doi.org/10.1007/s00339-019-2721-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-019-2721-7