
Abstract
Perovskite solid solution ceramics of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (BBSTBV, BBSTBT, 0.02 ≤ x ≤ 0.2) were prepared by the traditional solid state reaction technique. The phase evolution, microstructure and dielectric properties of BBSTBV and BBSTBT ceramics were researched. X-Ray diffraction results illustrated that both BBSTBV and BBSTBT could form a homogenous solid solution which has a similar structure with BaTiO3. The optimized properties of (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics with stable εr (~ 1769–2293), small Δε/ε25 °C values (± 15%) over a broad temperature range from − 58 to 151 °C and low tan δ ≤ 0.03 from − 11 to 131 °C were obtained. In the high-temperature region, the relaxation and conduction process are attributed to the thermal activation and the oxygen vacancies may be the ionic charge carriers in perovskite ferroelectrics.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Multilayer ceramic capacitors (MLCCs) are extensively used in electronic industry, which are closely related to our lives. Recently, some MLCCs are required to possess good temperature stability in a wide working temperature range owing to their work environments [1, 2]. The rapid increased working temperature of MLCCs induced that the EIA (the Electronic Industries Association) X7R (the change of the capacitance is less than 15% in the temperature range of − 55 to 125 °C) could not meet the requirements. So, the materials with excellent performance for X8R (the temperature range from − 55 to 150 °C) are developed [3].
Recently, as the main component of MLCCs, BaTiO3 has attracted much attention [4]. But pure BaTiO3 ceramics could not satisfy the requirements of temperature stability because of their phase transitions at ~ 5 and ~ 125 °C [5]. Therefore, many researchers are focused on the solid solutions between BT and Bi-based perovskite compounds [6], such as BaTiO3–Bi(Li0.5Zr0.5)O3 [7], BaTiO3–(Bi0.5Na0.5)TiO3 [8, 9], BaTiO3–Bi(Mg2/3Nb1/3)O3 [10], BaTiO3–Bi(Mg0.5Ti0.5)O3 [11] and so on. These systems exhibited good properties, showing that the modified barium titanate compositions are widely used in MLCCs due to their stable dielectric constant and the required temperature-stable dielectric behavior [12, 13].
In this paper, (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 ceramics were designed and prepared by the solid state reaction method. Furthermore, the phase evolution, microstructure, dielectric properties and impedance analysis of BBSTBV and BBSTBT ceramics were also systematically studied.
2 Experimental
(Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) samples were synthesized by the traditional solid state reaction technique as reported in our previous work [14]. The samples were sintered at 1200–1360 °C for 2 h in air. Dielectric properties were measured using a precision impedance analyzer (Model E4980AL, Hewlett-Packard Co, Palo Alto, CA) with an applied voltage of 500 mV over 100 Hz–1 MHz from − 150 to 200 °C at a heating rate of 2 °C/min. Other test instrument models are also described in our previous work [15].
3 Results and discussion
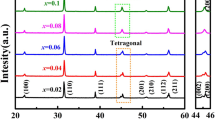
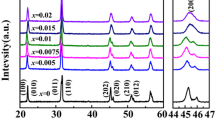
Figure 1 illustrates the XRD patterns of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) samples sintered at their optimized temperatures. No second phase was observed in Fig. 1, indicating that BBSTBV and BBSTBT have formed a homogenous solid solution. The enlarged XRD patterns in the range of 2θ from 44° to 46° are demonstrated in Fig. 1a2, b2. In Fig. 1a2, it is clearly seen that the samples with compositions of x ≤ 0.1 display an obvious splitting of (002)/(200) diffraction peaks, which correspond to the tetragonal symmetry. And the reducing intensities of (200) and (002) peaks could be clearly observed at around 45°. By contrast, the tetragonal characteristics of (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) could also be observed at 0.02 ≤ x ≤ 0.08. With further increasing the x values, the merging of (002)/(200) diffraction peak into (200) peak is illustrated in Fig. 1b2, which is the feature of pseudo-cubic, indicating that the tetragonal transformed into pseudo-cubic. Therefore, Ti4+ is easier to promote phase change than V5+ in the BaTiO3-based ceramics.

X-ray diffraction patterns of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 (a1) and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (b1) (0.02 ≤ x ≤ 0.2) ceramics, a2 and b2 are the enlarged XRD patterns of the samples in the range of 2θ from 44° to 46°
To clarify the phase evolution of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics, Raman spectra in the frequency range of 100–1000 cm− 1 at room temperature were carried out, the results are shown in Fig. 2. All samples demonstrated the existence of tetragonal or orthorhombic phases [16]. The negative Raman peak at 177 cm−1 for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 ceramics could be observed at 0.02 ≤ x ≤ 0.1 in Fig. 2a, which is the sign feature of the tetragonal [17]. When 0.02 ≤ x ≤ 0.08, the similar characteristic could be also obtained around 184 cm− 1 for (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 ceramics, as shown in Fig. 2b. Different bands at 177 and 304 cm−1, 184 and 306 cm−1 were the characteristic of long-range ferroelectric phase in Fig. 1a, b, respectively [18, 19]. The characteristics of the tetragonal and long-range ferroelectric vanished, pseudo-cubic phase appeared in its position with increasing x values. These results are consistent with the analysis of XRD patterns.

Room temperature Raman spectra of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics
Figure 3 illustrates the SEM images of the thermally etched surfaces for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 [(a) ~ (b)] and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 [(c) ~ (d)] (x = 0.1, 0.2) ceramics sintered at their optimized temperatures. Dense microstructure was observed in (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 samples as x = 0.1, as shown in Fig. 3a. With increasing x values, the grain size of the samples decreased slightly. For the compositions with the same x values, it could be clearly found that the grain size of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 samples was smaller than that of (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3, indicating that the ability of inhibiting for the grain growth of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 ceramics was better than (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 ceramics.

SEM micrographs of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 ceramics sintered at their optimized temperatures: a x = 0.1, 1280 °C and b x = 0.2, 1200 °C of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 samples, c x = 0.1, 1280 °C and d x = 0.2, 1200 °C of (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 samples
Figure 4 shows the Nyquist plots for the (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics with x = 0.2 at six temperatures. The Debye-like semicircles of both BBSTBV and BBSTBT ceramics became distorted and the center of the semicircles fell on below the real axis, demonstrating that the pure Debye-type relaxation is not observed in the dielectric response of all samples. With increasing the measured temperature, the impedance decreased, indicating that the conductivity of samples increased.

Nyquist plots of impedance for (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics at selected temperatures
Figure 5 represents the real (Z′) and imaginary (Z″) parts of the impedance with different frequency at six temperatures for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics with x = 0.2. As the frequency and temperature increased, the decrease of the Z′ and Z″ could be observed clearly. And the values of these two parts at different temperatures are combined separately in the high-frequency region, showing that the decrease in the barrier properties of the samples may lead to the release of the space charge. And it may be the dominant factor in the increase of ac conductivity for ceramics in high temperatures [20]. At lower frequency, higher impedance value is a characteristic of the space charge polarization of the materials, as demonstrated in Fig. 5a1, b1. In Fig. 5a2, b2, the peak of Z″ could be observed with increasing the frequency. Furthermore, the appearance of the peaks is a sign of the relaxation at different measured temperatures. The peak is asymmetric, and their positions move to high frequency with increasing measured temperatures. With increasing the measured temperature, the symmetry of the peak became worse and wider, indicating that the existence of electrical processes in the material with different relaxation times [21].

Frequency dependences of the real part Z′ (a) and imaginary part Z″ (b) of impedance for the (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics at selected temperatures [Insert shows the Arrhenius plot for relaxation time. The dotted line through the data is a linear fit to Eq. (2)]
The relaxation time (τ) of the (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics with x = 0.2 as a function of the absolute temperature is illustrated in the insert of Fig. 5a2, b2, respectively. In the relaxation system, the relaxation time (τ) can be calculated from the imaginary part (Z″) and frequency (f) curve of the impedance [Fig. 5]. The following relationship was used:
where fmax is the relaxation frequency. According to Eq. (1), the value of τ decreases with increasing the measured temperature, which indicates a typical semiconductor behavior. The activation energy (Ea) of this compound was calculated from Arrhenius relation:
where τ0, kB, Ea, and T are the preexponential factor, the Boltzmann constant, the activation energy required for the relaxation and the absolute temperature, respectively. From the slope of ln τ vs 103 T−1, the values of Ea are 1.51 and 1.64 eV for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics with x = 0.2, respectively.
Figure 6 demonstrates the frequency dependence of the AC conductivity (σac) for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics at six temperatures. With increasing the measured temperature, a dispersion of the conductivity could be observed. Meanwhile, the σac increased with increasing x values, and the σac of (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 is slightly bigger than that of (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 sample. This result is consistent with the analysis of the complex impedance. It is worth noting that the σac became independent of the frequency after reaching a certain value with decreasing the frequency. So, extrapolation of this part towards lower frequency could yield the direct-current conductivity. It could be expressed through the universal dielectric response law [22]:

Frequency dependences of ac conductivity for the (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 (a1) and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 (b1) ceramics at selected temperatures. a2, b2 are the Arrhenius plot of dc conductivity of the (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3, respectively. Squares are experimental points and the dotted line is a linear fit to Eq. (4)
where σdc is the dc conductivity, ω is the angular frequency of ac field. The A and n (0 < n < 1) are two temperature-dependent adjusting constants. The temperature dependence of the σdc could be obtained by the Arrhenius law:
where σ0 is the pre-exponential factor, Edc is the activation energy of conduction. Edc was calculated from the slope of ln(σ) vs 1000/T. The values are 1.44 and 1.81 eV for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics with x = 0. 2, respectively, as shown in Fig. 6a2, b2. These activation energies are approximately the same as the activation energies of relaxation, indicating that these two processes may be caused by the same factor of charge carriers, oxygen vacancies. In perovskite ferroelectric materials, oxygen vacancies are considered the one of the mobile charge carriers [23].
The oxygen vacancy could be produced from the substitution of B-site by mulitvalence V cation. Vanadium has multiple valences, such as V(0), V(III), V(IV), V(V), so X-ray photoelectron spectroscopic (XPS) analysis of the (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 sample was carried out to obtain the valence of V. Figure 7 shows the survey spectrum and V 2p high resolution XPS of the (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 sample, all peaks are calibrated with respect to C 1 s peak at 284.8 eV. There are XPS signals for Ba, Ti, Bi, Sr, V, O, and C elements. Kurmaev et al. [24] reported the BE values of V 2p3/2 (515.95 eV) and V 2p1/2 (523.48 eV) in the single crystal VO2. Biesinger et al. [25] reported the BE values of V 2p3/2 (517.20 eV) and V 2p1/2 (524.50 eV) in V2O5. Comparing with the V 2p spectra, the V oxidation states in (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 contained both V(IV) and V(V), as shown in Fig. 7b. The mass percentages of V element are 96.29% for V(IV) and 3.71% for V(V). Therefore, oxygen vacancies could be created. Besides, oxygen vacancy may be also obtained from the evaporation of Bi in high-temperature sintering [26]. The charge compensation follows reaction (Kröger and Vink) [27]:

Survey spectrum (a) and high resolution XPS of V 2p for (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 samples sintered at their optimized temperatures
The double charge oxygen vacancy is deemed to be the most mobile charge and has an important impact on conduction.
Figure 8 shows the temperature dependences of the relative permittivity (εr) and dielectric loss (tan δ) for the (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics at various frequencies in the measured temperature range from − 90 to 200 °C. With increasing x values, the peak of the relative permittivity depressed and became extremely broad at the Curie temperature, and the peak value decreased from ~ 7312 (x = 0.02, 1 kHz) to ~ 1906 (x = 0.1, 1 kHz), ~ 9068 (x = 0.02, 1 kHz) to ~ 2175 (x = 0.1, 1 kHz), respectively. For the relative permittivity, the value decreased with increasing frequency. The ∆ε/ε25 °C and tan δ of ceramics got the ideal optimization as x = 0.2. So, the ∆ε/ε25 °C, εr and tan δ of (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics at 1, 10 and 100 kHz from − 120 to 200 °C are illustrated in Fig. 9a, b, respectively. (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 samples with high εr (~ 1694–1906), small Δε/ε25 °C values (± 15%) over a broad temperature range from − 44 to 158 °C and low tan δ ≤ 0.03 from − 130 to 105 °C. (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 samples with high εr (~ 1769–2293), small Δε/ε25 °C values (± 15%) over a broad temperature range from − 58 to 151 °C and low tan δ ≤ 0.03 from − 11 to 131 °C were observed, indicating that the (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics may be an ideal candidates for MLCCs.

Temperature dependences of the relative permittivity and dielectric loss for (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics measured at 1, 10 and 100 kHz

∆ε/ε25 °C, Temperature dependence of relative permittivity, and dielectric loss of (Ba0.8Bi0.066Sr0.134)(Ti0.8Bi0.134V0.066)O3 and (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 ceramics from − 120 to 200 °C at 1, 10, 100 and 100 Hz, 1, 10 kHz, respectively
4 Conclusions
(Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 (0.02 ≤ x ≤ 0.2) ceramics have been prepared by the solid-state processing techniques. The phase evolution, microstructure and dielectric properties of BBSTBV and BBSTBT ceramics were investigated. The transformation of (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 sample from tetragonal to pseudo cubic phase occurred at 0.08 ≤ x ≤ 0.1. (Ba0.8Bi0.1Sr0.1)(Ti0.8Bi0.1Ti0.1)O3 samples exhibited good performances with high εr (~ 1769–2293), small Δε/ε25 °C values (± 15%) over a broad temperature range from − 58 to 151 °C and low tan δ ≤ 0.03 from − 11 to 131 °C. The relaxation and the conduction process in the high-temperature region are attributed to the thermal activation, and the oxygen vacancies may be the ionic charge carriers.
References
M.R. Werner, W.R. Fahrner, IEEE Trans. Ind. Electron. 48, 249–257 (2001)
A. Zeb, S. Milne, J. Mater. Sci. Mater. Electron. 26, 9243–9255 (2015)
L.X. Li, J.X. Chen, N. Zhang, Y.R. Liu, J.Y. Yu, J. Mater. Sci. Mater. Electron. 26, 84–89 (2015)
Y. Sakabe, Am. Ceram. Soc. Bull. 66, 1338–1341 (1987)
D. Hennings, G. Rosenstein, J. Am. Ceram. Soc. 67, 249–254 (1984)
Y.J. Wu, Y.Q. Lin, S.P. Gu, X.M. Chen, Appl. Phys. A Mater. Sci. Process. 97, 191–194 (2009)
X.L. Chen, X.X. Li, G.S. Huang, G.F. Liu, X. Yan, H.F. Zhou, J. Mater. Sci. Mater. Electron. 28, 17278–17282 (2017). https://doi.org/10.1007/s10854-017-7659-y
N. Zhang, L. Li, J. Chen, Y. Liu, J. Yu, Ceram. Int. 41, 4805–4809 (2014)
S.F. Wang, Y.F. Hsu, Y.W. Hung, Y.X. Liu, Appl. Sci. 5, 1221–1234 (2015)
X.L. Chen, J. Chen, L. Fang, D.D. Ma, G.F. Liu, H.F. Zhou, J. Am. Ceram. Soc. 98, 804–810 (2014)
J. Chen, X.L. Chen, F. He, Y.L. Wang, H.F. Zhou, L. Fang, J. Am. Ceram. Soc. 94, 4335–4339 (2011)
Q. Li, J.Q. Qi, Y.L. Wang, Z.L. Gui, L.T. Li, J. Am. Ceram. Soc. 21, 2217–2220 (2001)
W.H. Lee, C.Y. Su, J. Am. Ceram. Soc. 90, 3345–3348 (2007)
D.D. Ma, X.L. Chen, G.S. Huang, J. Chen, H.F. Zhou, F. Fang, Ceram. Int. 41, 7157–7161 (2015)
X.L. Chen, Y.L. Wang, J. Chen, H.F. Zhou, L. Fang, L.J. Liu, J. Am. Ceram. Soc. 96, 3489–3493 (2013)
C.H. Perry, D.B. Hall, Phys. Rev. Lett. 15, 700–702 (1965)
M.H. Frey, D.A. Payne, Phys. Rev. B 54, 3158–3168 (1996)
R. Farhi, M.E. Marssi, A. Simon, J. Ravez, Euro. Phys. J. B 9, 599–604 (1999)
A. Scalabrin, A.S. Chaves, D.S. Shim, S.P.S. Porto, Phys. Status Solidi B 79, 731–742 (1977)
J. Plocharski, W. Wieczoreck, Solid State Ion. 28, 979–982 (1988)
M. Boukriba, F. Sediri, N. Gharbi, Mater. Res. Bull. 48, 574–580 (2013)
A.K. Jonscher, J. Phys. D Appl. Phys. 32, R57-R70 (1999)
S. Sen, R.N.P. Choudhary, P. Pramanik, Phys. B 387, 56–62 (2007)
E.Z. Kurmaev, V.M. Cherkashenko, Y.M. Yarmoshenko, S. Bartkowski, A.V. Postnikov, M. Neumann et al., J. Phys. Condens. Matter 10, 4081–4091 (1998)
M.C. Biesinger, L.W.M. Lau, A.R. Gerson, R.S.C. Smart, Appl. Surf. Sci. 257, 887–898 (2010)
C.R. Zhou, X.Y. Liu, M.H. Jiang, C.L. Yuan, J. Cent. South. Univ. 41, 1796–1800 (2010)
F.A. Kröger, H.J. Vink, J. Phys. Chem. Solids 5, 208–223 (1956)
Acknowledgements
This work was supported by the Natural Science Foundation of China (nos. 11664008, 61761015, and 11464009), Natural Science Foundation of Guangxi (nos. 2017GXNSFDA198027, 2015GXNSFDA139033 and 2017GXNSFFA198011).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Chen, X., Li, X., Yan, X. et al. (Ba1−xBi0.33xSr0.67x)(Ti1−xBi0.67xV0.33x)O3 and (Ba1−xBi0.5xSr0.5x)(Ti1−xBi0.5xTi0.5x)O3 solid solutions: phase evolution, microstructure, dielectric properties and impedance analysis. Appl. Phys. A 124, 423 (2018). https://doi.org/10.1007/s00339-018-1833-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00339-018-1833-9