
Abstract
Linkage disequilibrium (LD) affects genomic studies accuracy. High-density genotyping platforms identify SNPs across animal genomes, increasing LD evaluation resolution for accurate analysis. This study aimed to evaluate the decay and magnitude of LD in a cohort of 81 crossbred dairy cattle using the GGP_HDv3_C Bead Chip. After quality control, 116,710 Single Nucleotide Polymorphisms (SNPs) across 2520.241 Mb of autosomes were retained. LD extent was assessed between autosomal SNPs within a 10 Mb range using the r2 statistics. LD value declined as inter-marker distance increased. The average r2 value was 0.24 for SNP pairs < 10 kb apart, decreasing to 0.13 for 50–100 kb distances. Minor allele frequency (MAF) and sample size significantly impact LD. Lower MAF thresholds result in smaller r2 values, while higher thresholds show increased r2 values. Additionally, smaller sample sizes exhibit higher average r2 values, especially for larger physical distance intervals (> 50 kb) between SNP pairs. Effective population size and inbreeding coefficient were 150 and 0.028 for the present generation, indicating a decrease in genetic diversity over time. These findings imply that the utilization of high-density SNP panels and customized/breed-specific SNP panels represent a highly favorable approach for conducting genome-wide association studies (GWAS) and implementing genomic selection (GS) in the Bos indicus cattle breeds, whose genomes are still largely unexplored. Furthermore, it is imperative to devise a meticulous breeding strategy tailored to each herd, aiming to enhance desired traits while simultaneously preserving genetic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
When alleles are sufficiently close together on a chromosome, they tend to be inherited together through linked inheritance rather than being passed down independently. This means that the offspring receives blocks of alleles or haplotypes from each parent, rather than individual alleles (O’Brien et al. 2014; Ardlie et al. 2002). Linkage disequilibrium (LD) arises due to the interconnection of alleles on a chromosome, which results in a certain degree of correlation among them. This phenomenon can be observed not only for nucleotides in the genome but also for different types of genetic markers, including single nucleotide polymorphisms (SNPs) (O’Brien et al. 2014). In essence, LD between molecular markers represents the extent to which the genotypes of two SNPs are correlated (Porto-Neto et al. 2014). While physical proximity plays a significant role in determining LD, it is important to acknowledge that other factors such as evolutionary processes and historical events can also affect the correlation between molecular markers. These factors encompass inbreeding, selection, population stratification, genetic drift, genetic bottleneck, effective population size, mutation, recombination rate, and migration (Karimi et al. 2015; Ardlie et al. 2002; Reich et al. 2001).
Comprehending the concept of LD holds paramount importance in the mapping of genes and their relevance to genomic studies. Through the analysis of LD between SNPs, researchers can obtain valuable insights into the diversity across various breeds, estimate recombination event frequencies, investigate fluctuations in effective population size across generations, and identify genomic regions suitable for enhancing economically important traits. Such understanding contributes significantly to the advancement of genetic research and the targeted improvement of desired traits (O’Brien et al. 2014; Espigolan et al. 2013; McKay et al. 2007).
Effective population size is an important genetic parameter that estimates the effect of genetic drift in a population (Crow and Kimura 1970). It is one of the best quantitative indicators of genetic diversity (Makanjuola et al. 2020), that help determine the number of independent chromosome segments which are required for genomic predictions (Mrode et al. 2019). Estimating the effective population size is valuable not only from an evolutionary standpoint but also for enhancing models used in the mapping of genes related to quantitative traits (Li and Kim 2015).
The decreasing cost of conducting high-throughput genotyping assays has opened up new possibilities for conducting large-scale genomic studies. The effectiveness and precision of these genomic investigations are greatly influenced by the extent and structure of LD observed between SNPs throughout the genome. As LD patterns determine the level of correlation between markers, understanding LD is crucial for maximizing the utility and accuracy of GWAS and GS approaches (Goddard and Hayes 2012). Investigations have been carried out to examine the LD between markers within the genomes of diverse taurine and indicine cattle breeds (Espigolan et al. 2013; Makina and Taylor 2015). The findings of their study showed that moderate LD with a value of r2 = 0.20 was observable in distances of less than 100 kilobases. This suggests that a set of 50,000 SNPs is sufficient to capture most of the LD information needed for conducting GWAS in taurine breeds (McKay et al. 2007; Espigolan et al. 2013). In contrast, the researchers observed a lower extent of LD (r2 = 0.20–0.34) within indicine cattle at distances less than 30 kilobases. This suggests that a higher-density SNP chip would be necessary to capture the LD information required for conducting genomic studies in these cattle (Makina and Taylor 2015). The diminished LD extent observed in indicine cattle breeds can be attributed to a potential ascertainment bias inherent in the SNP chips utilized for genotyping.
The practice of crossbreeding in dairy cows has been identified as a highly effective approach for enhancing livestock productivity, reproductive efficiency, and sustainability (Leroy et al. 2016; Mbole-Kariuki et al. 2014; Bebe et al. 2003). In a single lactation, the crossbred offspring of Sahiwal (Bos indicus) and HF (Bos taurus) cows have the ability to produce approximately 4000 L of milk containing 4% fat (Kumar et al. 2018). Crossbreeding Sahiwal and HF cows result in offsprings that benefit from hybrid vigor, which leads to improved health traits and increased productivity. This is due to the combination of the high milk yield of HF cows and the adaptability and heat tolerance of Sahiwal cows, resulting in offsprings that are well-suited to local conditions and have improved milk yield.
In a recent study, admixture patterns and signature of selection for the same samples were studied (unpublished data). However, the investigation of genome-wide LD and its pattern using this specific chip remains unexplored. The prediction accuracy from genomic selection (GS) is affected by marker density, minor allele frequency (MAF), and genetic architecture of the target trait (Zhang et al. 2019). Also, the accuracy of the genomic prediction depends on the amount of genetic variation explained by the markers resulting from the LD between the marker and QTL (Goddard 2009). Therefore, this study examines the distribution of allelic frequencies, determines the level of LD (measured using r2), and estimates the effective population size in the population of crossbred dairy cattle, which have a major impact on the accuracy of GS in this admixed population. This study will provide valuable insights for estimating marker density in genomic studies of crossbred dairy cattle.
Materials and methods
Ethics statement
To ensure the ethical and humane treatment of animals, the study described in this research paper was approved by the institutional review committee. During blood collection, a professional veterinarian was there to ensure minimal distress and harm to the animals. Before collecting any samples, the researchers met with the owners of the farm where the animals were housed to explain the purpose of the study and obtain informed consent verbally.
Animal sample and genotype quality control
The sample size for this study consisted of 81 crossbred cattle from the Military Farm located in Renala Khurd near Okara, Punjab. These animals were selected based on having varying percentages of HF and Sahiwal genetics from different lactations. Due to the nature of crossbreeding, the breed composition varies from individual to individual due to variations in Sahiwal and HF inheritance. For instance, some crossbreds have an approx. 50% inheritance from both HF and Sahiwal, while others have 31/32 parts of HF inheritance the remaining being Sahiwal. Blood sampling was carried out in different visits to cattle farms during 2021 and 2022.
DNA was extracted from the blood samples using the FavorPrep™ Blood Genomic DNA Extraction Mini Kit, following the manufacturer's guidelines. The quality and quantity of the DNA were evaluated using different methods, including a NanoDrop spectrophotometer, agarose gel electrophoresis, and a Qubit spectrophotometer. The extracted DNA was genotyped using the GGP_HDv3_C (GeneSeek® Genomic Profiler™) and commercially available services at GeneSeek (Neogen Corporation, Lincoln, NE, United States). The genotypes were identified and analyzed using the Genome Studio software from Illumina, Inc. The analysis was based on the bovine genome assembly, ARS-UCD1.2.
Quality control (QC)
After the genotyping process, the initial raw data comprised 139,376 SNPs for the crossbred individuals. Quality control measures were applied using the PLINK v1.9 software (Slifer 2018) which involved removing SNPs that had a call rate of less than 95%, minor allele frequency (MAF) of less than 0.02, and a Hardy–Weinberg equilibrium (HWE) of less than 10E−05. For subsequent analysis, only autosomal SNPs were considered.
Marker statistics
The R software was employed to estimate multiple characteristics of the autosomes. These included the length of each chromosome in megabases (Mb), the count of markers on each autosome, the longest and shortest intervals between SNPs, and the average interval between SNPs across all autosomes (Team 2020).
Minor allele frequency (MAF)
To calculate the MAF of autosomal SNPs, default settings in PLINK v1.9 software were used with the command “--file data --freq” (Slifer 2018). The distribution of allele frequencies across various chromosomes was analyzed using the R software. Additionally, a plot was generated to visualize the proportion of SNPs falling within different frequency categories, namely 0.02–0.10, 0.10–0.20, 0.20–0.30, 0.30–0.40, and 0.40–0.50 (Team 2020).
Inbreeding coefficient (F) and effective population size (Ne)
To estimate F, the expected and observed homozygote differences were used with PLINK v1.9 software (Slifer 2018) using the formula Fi = (Oi − Ei)/Li − Ei. In this equation, Fi is the estimated inbreeding coefficient of the ith animal, Oi represents the number of observed homozygous loci, Ei represents the count of expected homozygous loci, while Li represents the count of genotyped autosomal loci. The calculation was performed using PLINK v1.9 software (Slifer 2018).
To estimate the effective population size (Ne), the SNeP tool was utilized, leveraging the relationship between Ne, linkage disequilibrium (represented by r2), and recombination rate (c) (Barbato et al. 2015). This is given by Corbin et al. (2012):
In the provided equation: NT(t) represents the estimated effective population size t generations ago in the past, Ct denotes the recombination rate t generations ago in the past, r2adj signifies the adjusted LD estimation, accounting for sampling bias, f is mapping function and α represents a constant value.
Linkage disequilibrium (LD)
The assessment of LD was carried out using the square of the correlation coefficient between two loci, represented as r2. This metric is regarded as robust and unaffected by fluctuations in allele frequency and population size (Zhao et al. 2007). The estimation of r2 plays a crucial role in determining the number of loci needed for conducting GWAS and quantitative trait loci (QTL) mapping. This measure helps to assess the extent of LD between markers and assists in designing the appropriate sample size and marker density for such studies (Makina and Taylor 2015). The equation for estimating LD using the r2 value is expressed as follows:
In this context, the frequency of the ith allele at locus A is denoted as pAi, while the frequency of the jth allele at locus B is represented as pBj. Additionally, the frequency of the haplotype AiBj in the population is denoted as pAiBj.
The MapThin v1.11 was used to thin the map files, selecting 20 SNPs per 106 bp positions to minimize false positive results and increase the efficiency of the analysis (Howey and Cordell 2011). The PLINK v1.9 software was used with the default command “--ld-snp-list mysnplist --ld-window-kb 186,000 --ld-window 99,999 --ld-window r2 0” to estimate the r2 between all pairs of SNPs on autosomes as the length of the longest chromosomes (chr1) is around 186 kb (Slifer 2018).
To verify the occurrence of free recombination at a physical distance greater than 10 Mb, two types of analyses were conducted on our dataset: one without considering a 10 Mb window, and the other with considering a 10 Mb window (Zhao et al. 2014).
-
1.
To analyze the decay of LD value, the genomic regions were divided into eight categories based on a range of 20 Mb each, namely 0–20 Mb, 20–40 Mb, 40–60 Mb, 60–80 Mb, 80–100 Mb, 100–120 Mb, 120–140 Mb, and 140–160 Mb. The LD value was then calculated for all possible regions within each category.
-
2.
By taking into account a maximum distance of 10 Mb between SNP pairs, the LD decay was calculated for all possible SNP pairs across the autosomes. The trend in Linkage Disequilibrium (LD) decay for crossbred individuals was plotted across the entire first 10 megabases (MB) of the genome. This analysis likely provides insights into how LD changes over increasing physical distances within this specific genomic region for crossbred animals.
The calculated LD decay was then categorized into eight intervals based on distance ranges. These intervals included: 0–10 kb, 10–25 kb, 25–50 kb, 50–100 kb, 100–500 kb, 0.5–1 Mb, 1–5 Mb, and 5–10 Mb. This categorization allows for a comprehensive assessment of LD decay patterns across varying distances, providing valuable insights into the dynamics of LD in the autosomal genome and plotted against distance range.
Minor allele frequency and sample size impact
To assess the impact of minor allelic frequency (MAF) and sample size on LD, the analysis was extended. For a physical distance of 10 Mb, LD was computed using four distinct MAF thresholds (0.05, 0.10, 0.15, and 0.2). Furthermore, seven random subsets of the population were selected with different sample sizes (N = 10, 20, 30, 40, 50, 60, and 70) to investigate the impact of sample size on r2-based LD. The extent of LD was assessed for each subset, and the impact of sample size and MAF on LD (r2) was also depicted through plotting.
Results
Quality control
Figure 1 summarizes the quality control results for different MAF thresholds. For example, for a 0.02 MAF threshold, 1804 SNPs were removed due to an MAF less than 0.02; 216 SNPs were removed based on Hardy–Weinberg Equilibrium (HWE); and 1111 SNPs were excluded due to a call rate threshold criterion. Therefore, a total of 116,710 autosomal SNPs with a genotypic rate of 0.99 were available for downstream analysis. These steps were repeated for different MAF values. All the remaining parameters were the same therefore the effect of different maf values on the final number of SNPs left for downstream analysis is depicted in Fig. 1.

Effect of different MAF thresholds on the total no. of SNPs left for downstream analysis
Marker statistics
The quality control process resulted in a total of 2520.241 Mb of retained SNPs across the genome of crossbred dairy cattle, with an average chromosome length of 86.90 Mb. The longest chromosome was BTA1, with a length of 158.8551 Mb, while the shortest was BTA25, with a length of 42.85 Mb. The number of SNPs on each chromosome exhibited a proportional relationship with the length of the respective chromosome. Notably, the highest number of SNPs was observed on BTA1 (7078), while the lowest number was recorded on BTA25 (1945). On average, the distance between adjacent SNPs was approximately 21.70 kb. The longest distance between SNPs was observed on BTA5 (612 kb), and the longest distance between SNPs on the same chromosome was found on BTA5 (3882 kb). Conversely, the mean shortest distance between SNPs was 0.16 kb, with the shortest distance occurring on BTA18 (0.002 kb). Descriptive statistics for each autosome's SNP markers are provided in Table 1.
Minor allele frequency (MAF)
The mean MAF observed across all autosomes was recorded as 0.30. Figure 2 provides a visual representation of the distribution of MAF on all autosomes.

Minor allele frequency (MAF) in all autosomes
Similarly, the distribution of MAF indicated that a significant percentage of SNPs exhibited elevated MAF values (Fig. 3). Specifically, around 54% of the SNPs were categorized in the last two MAF groups (MAF ≥ 0.3), while a lower percentage of SNPs fell into the initial categories. On average, around 25.20% of the SNPs displayed an MAF value lower than 0.2. This distribution highlights the predominance of SNPs with higher MAF values in the analyzed dataset.

Proportion of SNPs categorized by minor allele frequencies (MAF) across autosomal chromosomes
It is noteworthy that all autosomes exhibited a similar trend, with a greater percentage of SNPs falling into the last two groups (MAF ≥ 0.3). However, BTA25, BTA6, BTA22, BTA9, and BTA23 had a higher percentage of SNPs in the last category (MAF ≥ 0.4). Among the chromosomes, BTA19 (10.04%), BTA17 (9.67%), BTA4 (9.61%), and BTA14 (9.54%) had a higher percentage of SNPs with MAF values between 0.02 and 0.1.
Inbreeding coefficient (F) and effective population size (Ne)
The average value of F was estimated to be 0.028, indicating that the risk of negative impacts due to inbreeding depression can be considered insignificant at this level of inbreeding.
The Ne of the crossbred dairy cattle was estimated throughout the past 1000 generations based on the average r2 values, as presented in Table 2. The results showed a declining trend in Ne, which decreased from 2775 (995 generations ago) to 150 (13 generations ago). This suggests that the crossbred dairy cattle population has experienced a decrease in genetic diversity over time.
Extent of LD across the genome
Without considering the window, we obtained a total of 47,054,338 possible pairs in the whole dataset, with a mean r2 value of 0.020025 (Table 3). While a total of 9,115,588 combination pairs across the autosomes were analyzed to estimate LD for SNP pairs with a physical distance of ≤ 10 Mb. The mean r2 value for markers at a 10 Mb distance was determined to be 0.128. Table 3 provides the mean LD (r2) values for different intervals of physical distance.
Table 3 shows that considering a 10 Mb window is important because there is an inverse relationship between LD (r2) and the distance between SNP pairs. The mean r2 values are higher when the SNP pair distance is smaller, i.e., between 0 and 10 kb, and decrease from 0–10 kb to 5–10 Mb. Therefore, it is suggested that SNP pairs within a distance of 10 Mb should be explored further.
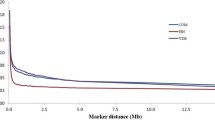
The level of Linkage Disequilibrium (LD) decay, about the distance between pairs of Single Nucleotide Polymorphisms (SNPs), is depicted in Fig. 4 for all autosomes. Notably, higher levels of LD were predominantly observed at shorter distances between SNP pairs, highlighting the rapid decay of LD as the physical distance between SNPs increases.

Average linkage disequilibrium (LD) decay in relation to the SNP pair distance
The level of LD measured by r2 varied across each chromosome, and was dependent on the physical distance between genetic markers. To explore the relationship, the mean r2 was computed for various physical distance intervals of markers across each chromosome. Chromosomes BTA22, BTA19, BTA18, and BTA7 exhibited higher levels of LD. When considering markers separated by < 10 kb, the average r2 was found to be 0.2332, which decreased to 0.1792 for markers with distances between 25 and 50 kb. The average r2 continued to decline with increasing distance, reaching a final value of 0.0344 for the 5–10 Mb category. These results indicate that the mean r2 values decrease as the physical distance between markers increases, demonstrating a decline in LD with increasing genetic distance (Fig. 5).

Box plot of mean r2 and SNP Pair distance up to 10 Mb for all 29 autosomes
The average r2 values showed a significant difference across various autosomes, especially for SNP distances less than 10 kb. On the other hand, for SNP distances greater than 100 kb, lower mean r2 values with relatively little variation across different autosomes were observed (Fig. 5).
Minor allele frequency (MAF) and linkage disequilibrium (LD) estimates
The impact of MAF on the magnitude of LD was examined by employing four threshold levels: 0.05, 0.10, 0.15, and 0.20. This analysis focused on SNP pairs with 10 Mb physical distances (Fig. 6). The findings revealed a notable influence of the MAF threshold on the average r2, especially in the case of shorter distances between SNPs. A decrease in the r2 value between SNP pairs was observed when the MAF threshold was set to a lower value (0.05), while a substantial increase in the r2 value was noted at higher thresholds of MAF. The mean r2 values ranged from 0.03 to 0.25 for MAF > 0.05, 0.03 to 0.27 for MAF > 0.10, 0.03 to 0.32 for MAF > 0.15, and 0.04 to 0.34 for MAF > 0.20. These results indicated that the MAF threshold has a considerable effect on the LD extent between SNPs, with higher MAF thresholds resulting in stronger LD.

Effect of minor allele frequency (MAF) on linkage disequilibrium extent
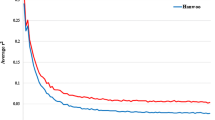
Sample size and LD estimates
To study the effect of sample size, random samples of sizes 10, 20, 30, 40, 50, 60, and 70 were selected from the total population for analysis. A noteworthy finding of this study is that the average r2 increased for smaller sample sizes, particularly when the physical distance intervals between SNP pairs exceeded 50 kb (Fig. 7). These results suggest that a larger sample size (at least 40 animals) is needed for an accurate estimation of r2.

Effect of different sample size on mean r2 estimates
Discussion
Following rigorous quality control measures, a final set of 116,710 autosomal SNPs were retained for analysis. These SNPs spanned a genomic region of 2520.241 Mb in crossbred dairy cattle. The average MAF was found to be 0.30, aligning with previously reported MAF values observed in diverse cattle breeds (Makina and Taylor 2015). These findings align with previous studies on other taurine breeds of cattle (O’Brien et al. 2014; Matukumalli et al. 2009; McKay et al. 2007). However, the average MAF values observed in this study were notably higher compared to indicine breeds of cattle, which typically exhibit MAF values ranging from 0.19 to 0.20 (O’Brien et al. 2014; Espigolan et al. 2013; Silva et al. 2010). In contrast to taurine cattle, indicine breeds typically display a distinct pattern in MAF levels, characterized by a greater representation of alleles with lower frequencies (< 0.2) (O’Brien et al. 2014; Gibbs et al. 2009; Villa-Angulo et al. 2009). The variation mentioned above could be ascribed to the increased genetic diversity identified in indicine breeds (Murray et al. 2010; Gibbs et al. 2009). As there are few such reports on crossbred cattle, especially with Bos Indicus and Bos taurus crossbreds, this may serve as a positive contribution and important reference for the future studies of native breeds. Furthermore, the commercially available SNP panel used in this study predominantly utilized sequence data from Bos taurus breeds. Thus, it may lead to the ascertainment bias leading to a greater proportion of SNPs with low MAF in indicine breeds of cattle.
The distribution of MAF has a direct impact on the extent of LD, as a lower MAF can result in a greater difference in allelic pair frequencies, leading to an underestimation of LD (Wray 2005). To examine the effect of MAF on LD, four different MAF thresholds were selected. The results indicated that higher thresholds of MAF (> 0.20) were associated with higher average LD (r2) between SNPs, particularly at shorter distances (O’Brien et al. 2014; Sargolzaei et al. 2008; Uimari et al. 2005). At a lower MAF threshold (e.g., 0.05), there might be the inclusion of rare variants in the analysis. Rare variants may behave differently in terms of LD leading to the observed decrease in r2 as they are less likely to be in strong linkage with other variants. Although, it has been established till now that the r2 method is less affected by the sample size it may also be a contributing factor to this finding indicating that SNP chips with a higher density of SNPs and studies on larger populations may be preferable for genomic studies in native cattle breeds of Pakistan.
The estimation of LD between SNP pairs was conducted using the correlation (r2) method, which is known to be less affected by MAF (Ardlie et al. 2002) and small sample size (Zhao et al. 2014). To assess the decay of LD, the physical distance between markers was divided into distinct intervals. The findings demonstrated a swift decrease in r2 beyond a threshold of 100 kb. Furthermore, r2 declined from 0.24 to 0.17 when considering marker distances of 10 kb and 50 kb, respectively. For inter-marker distances of up to 25 kb, the average r2 value was 0.24, which is comparatively lower than previous LD estimates, documented for taurine breeds such as Angus (0.46) and Hereford (0.49), as well as indicine breeds like Brahman (0.25) and Nellore (0.27) cattle (Porto-Neto et al. 2014; Espigolan et al. 2013; Lu et al. 2012). The results showed higher mean r2 values for BTA22, BTA19, BTA18, and BTA7, while lower mean r2 values were observed for BTA27, BTA21, BTA9, and BTA26.
LD (r2) values that surpass 0.3 are deemed valuable for dependable association studies and precise genomic predictions (Ardlie et al. 2002; Meuwissen et al. 2001, Kruglyak 1999). In this study, regions up to 10 kb on BTA5, BTA7, BTA13, BTA18, BTA22, BTA26, and BTA27 exhibited r2 values larger than 0.3. On the other hand, BTA19 showed a slower decay in LD, achieving the same level of r2 up to a distance of 25 kb. These findings deviate from the average LD levels documented in other taurine breeds such as Angus, Holstein, Brown Swiss, and Fleckvieh reach an average r2 value of 0.3 at distances ranging from 40 to 50 kb (O’Brien et al. 2014). These results align with the findings observed in indicine breeds such as Gyr and Nelore, which exhibit a more rapid decline in LD, reaching a similar r2 value at distances of approximately 20 kb. Similar outcomes have been observed in other taurine breeds, where r2 values remained comparable for distances equal to or less than 30 kb (Larmer et al. 2014; Bolormaa et al. 2011). Nevertheless, no correlation was identified between chromosomal size and r2 estimates (Bohmanova et al. 2010).
The LD decay analysis within a range of up to 10 megabases (MB), employing 10-kilobase (kb) windows, is presented in Fig. 4 for crossbred individuals. This analysis revealed a pronounced decline in LD at shorter distances between pairs of SNPs. This behavior is likely attributed to the relatively low number of SNP pair comparisons available at these close distances (Fig. 7).
These findings suggest that, when utilizing the set of SNPs contained in the GGPHDv3-C chip, there may not be consistent LD levels expected for genomic distances less than 10 MB. Importantly, these results align with those reported by O’Brien et al. (2014) in their LD analysis conducted across different taurine and indicine breeds.
To examine the influence of sample size on the extent of LD, various sample sizes were employed in the computation of r2 values as it is previously reported that a small sample size may lead to overestimation of LD (Yan et al. 2009; Khatkar et al. 2008). In the present investigation, a sample size of 40 cattle did not influence r2, which aligns with previous findings (Bohmanova et al. 2010; Singh et al. 2021). However, various other studies have reported different threshold limits for sample size. For example, Zhu et al. (2013) suggested a minimum sample size of 100. In the case of Holstein cattle a minimum sample size of 400 is necessary for reliable LD decay analysis (Khatkar et al. 2008). Human studies have indicated even higher sample sizes (Chen et al. 2006). The minimum threshold for sample sizes appears to be around 75, as r2 accuracy is significantly compromised below this value (Khatkar et al. 2008). Similarly, another study suggested a minimum sample size of 55 (Bohmanova et al. 2010).
Previous studies have consistently emphasized the significance of employing a larger number of SNPs to adequately cover the genome in genomic evaluations, especially when analyzing data from crossbreds and indicine breeds (Makina and Taylor 2015; Espigolan et al. 2013). The findings from our study support this notion, that a higher density SNP array provides more information and enhances the reliability of GWAS and GS in crossbred dairy cattle populations. These results are consistent with other studies that have also emphasized the benefits of utilizing a higher SNP density for such analyses (Singh et al. 2021).
To gain a better understanding of the population diversity and structure, we estimated F and Ne. Our study revealed an inbreeding coefficient of 0.028. This finding is comparable to previously reported inbreeding coefficients of 3%, 4%, and 6% in the Vrindavani crossbred cattle population of India (Chhotaray et al. 2021; Singh et al. 2021; Elavarasan et al. 2023). The Karan Fries crossbred cattle of Karnal exhibited an inbreeding coefficient of 3.68% (Mumtaz et al. 2021). In the case of Sahiwal cattle, one of the 11 breeds studied by Bang et al. (2022), the inbreeding coefficient was 0.9%. In Tharparker cattle, different methods were employed to estimate genomic inbreeding coefficients, resulting in values of 0.0589 (FROH), 0.0215 (FHOM), 0.0532 (FGRM), and 0.0160 (FUNI) (Saravanan et al. 2022). Another study was conducted on Holstein, Montebeliarde, and Normande breeds, an inbreeding coefficient of 4.5–5% (Dezetter et al. 2015). For pure African taurine (Baoulé) and its crossbreeds with indicine Zebu cattle, genomic inbreeding coefficients ranged from 0 to 4% (Ouédraogo et al. 2021). Among nine breeds, the mean genomic inbreeding estimates were highest for Jersey (0.173) and lowest for Hereford (0.051) (Kelleher et al. 2017). Lower inbreeding was observed in six Columbian cattle breeds, ranging from 0.5 to 4.5% (Martinez et al. 2023). In a study of 171 cattle groups conducted by Tian et al. (2023), the average inbreeding coefficient ranged from 0.22 to 0.05.
In our study, we observed a decrease in effective population size (Ne) over multiple generations in crossbred dairy cattle. The estimated Ne in our population was 150, and a decreasing trend in Ne was observed specifically 13 generations ago. When the effective population size (Ne) decreases, the genetic diversity available for selection in genomic breeding is constrained. This reduction in diversity limits the number of allelic variants that can be considered. Additionally, increased inbreeding resulting from a smaller Ne compromises fitness and undermines the accuracy of predictions due to the correlation of genetic variants. Moreover, the limited genetic contributions and heightened genetic drift further hinder the effectiveness of GS. When comparing our findings to previous studies, a range of 33–153 Ne was observed in different studies on dairy cattle (Doekes et al. 2018; Rodríguez-Ramilo et al. 2015; Stachowicz et al. 2011). Effective population sizes for European taurine breeds ranged from 98 to 152, with Brown Swiss exhibiting the lowest value (98), and Limousine and Piedmontese showing the highest values (138 and 144, respectively). African taurine breeds recorded a range of 120–175 for Ne. Among indicine cattle breeds, Gir had the highest Ne estimate (180), while Tharparker had the lowest (63) (Barbato et al. 2020). In the context of buffalo breeds, both purebred and crossbred populations demonstrated a decreasing trend in recent Ne, with estimated values closer to 387 and 113, respectively, 13 generations ago. This suggests that these animals have undergone strong selection or genetic drift, resulting in a decline in population size (Deng et al. 2019).
Conclusion
This study aimed to assess the level of LD between markers in crossbred dairy cattle from Pakistan using the GGP_HDv3_C (GeneSeek® Genomic Profiler™) SNP panel. The average estimated value of r2 was 0.24, which was lower compared to both indicine and taurine breeds. This suggests that a denser SNP panel is necessary to obtain more precise and accurate results in whole genome association studies for crossbred dairy cattle.
Additionally, the study observed a declining trend in the estimates of effective population size (Ne) in the population. This indicates the need for a well-designed breeding plan that can maintain a sufficiently large Ne to mitigate the negative effects of genetic drift and inbreeding.
Conducting studies on a larger population using a high-density array of SNPs would provide more comprehensive and reliable information regarding the extent of LD and the effective population size in crossbred dairy cattle of Pakistan.
Availability of data and materials
All the necessary files are provided with the paper.
References
Ardlie KG, Kruglyak L, Seielstad M (2002) Patterns of linkage disequilibrium in the human genome. Nat Rev Genet 3:299–309
Bang NN, Hayes BJ, Lyons RE, Randhawa IA, Gaughan JB, McNeill DM (2022) Genomic diversity and breed composition of Vietnamese smallholder dairy cows. J Anim Breed Genet 139:145–160
Barbato M, Orozco-Terwengel P, Tapio M, Bruford MW (2015) SNeP: a tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet 6:109
Barbato M, Hailer F, Upadhyay M, del Corvo M, Colli L, Negrini R, Kim E-S, Crooijmans RP, Sonstegard T, Ajmone-Marsan P (2020) Adaptive introgression from indicine cattle into white cattle breeds from Central Italy. Sci Rep 10:1–11
Bebe BO, Udo HM, Rowlands GJ, Thorpe W (2003) Smallholder dairy systems in the Kenya highlands: breed preferences and breeding practices. Livest Prod Sci 82:117–127
Bohmanova J, Sargolzaei M, Schenkel FS (2010) Characteristics of linkage disequilibrium in North American Holsteins. BMC Genomics 11:1–11
Bolormaa S, Hayes B, Savin K, Hawken R, Barendse W, Arthur P, Herd R, Goddard M (2011) Genome-wide association studies for feedlot and growth traits in cattle. J Anim Sci 89:1684–1697
Chen Y, Lin C-H, Sabatti C (2006) Volume measures for linkage disequilibrium. BMC Genet 7:1–8
Chhotaray S, Panigrahi M, Pal D, Ahmad SF, Bhanuprakash V, Kumar H, Parida S, Bhushan B, Gaur G, Mishra B (2021) Genome-wide estimation of inbreeding coefficient, effective population size and haplotype blocks in Vrindavani crossbred cattle strain of India. Biol Rhythm Res 52:666–679
Corbin LJ, Liu A, Bishop S, Woolliams J (2012) Estimation of historical effective population size using linkage disequilibria with marker data. J Anim Breed Genet 129:257–270
Crow J, Kimura M (1970) An introduction to population genetics theory. Harper & Row, New York
Deng T, Liang A, Liu J, Hua G, Ye T, Liu S, Campanile G, Plastow G, Zhang C, Wang Z (2019) Genome-wide SNP data revealed the extent of linkage disequilibrium, persistence of phase and effective population size in purebred and crossbred buffalo populations. Front Genet 9:688
Dezetter C, Leclerc H, Mattalia S, Barbat A, Boichard D, Ducrocq V (2015) Inbreeding and crossbreeding parameters for production and fertility traits in Holstein, Montbéliarde, and Normande cows. J Dairy Sci 98:4904–4913
Doekes HP, Veerkamp RF, Bijma P, Hiemstra SJ, Windig JJ (2018) Trends in genome-wide and region-specific genetic diversity in the Dutch-Flemish Holstein-Friesian breeding program from 1986 to 2015. Genet Sel Evol 50:1–16
Elavarasan K, Kumar S, Agarwal S, Vani A, Sharma R, Kumar S, Chauhan A, Sahoo NR, Verma MR, Gaur GK (2023) Estimation of microsatellite-based autozygosity and its correlation with pedigree inbreeding coefficient in crossbred cattle. Animal Biotechnol 2023:1–14
Espigolan R, Baldi F, Boligon AA, Souza FR, Gordo DG, Tonussi RL, Cardoso DF, Oliveira HN, Tonhati H, Sargolzaei M (2013) Study of whole genome linkage disequilibrium in Nellore cattle. BMC Genomics 14:1–8
Gibbs RA, Taylor JF, van Tassell CP, Barendse W, Eversole KA, Gill CA, Green RD, Hamernik DL, Kappes SM (2009) Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324:528–532
Goddard M (2009) Genomic selection: prediction of accuracy and maximisation of long term response. Genetica 136:245–257
Goddard M, Hayes B (2012) Genome-wide association studies and linkage disequilibrium in cattle. Bovine Genomics 2012:192–210
Howey R, Cordell H (2011) MapThin. http://www.staff.ncl.ac.uk/richard.howey/mapthin/
Karimi K, Esmailizadeh Koshkoiyeh A, Gondro C (2015) Comparison of linkage disequilibrium levels in Iranian indigenous cattle using whole genome SNPs data. J Animal Sci Technol 57:1–10
Kelleher M, Berry D, Kearney J, McParland S, Buckley F, Purfield D (2017) Inference of population structure of purebred dairy and beef cattle using high-density genotype data. Animal 11:15–23
Khatkar MS, Nicholas FW, Collins AR, Zenger KR, Cavanagh JA, Barris W, Schnabel RD, Taylor JF, Raadsma HW (2008) Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics 9:1–18
Kruglyak L (1999) Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genetics 22:139–144
Kumar S, Alex R, Gaur G, Mukherjee S, Mandal D, Singh U, Tyagi S, Kumar A, Das A, Deb R (2018) Evolution of Frieswal cattle: a crossbred dairy animal of India. Indian J Anim Sci 88:265–275
Larmer S, Sargolzaei M, Schenkel F (2014) Extent of linkage disequilibrium, consistency of gametic phase, and imputation accuracy within and across Canadian dairy breeds. J Dairy Sci 97:3128–3141
Leroy G, Baumung R, Boettcher P, Scherf B, Hoffmann I (2016) Sustainability of crossbreeding in developing countries; definitely not like crossing a meadow…. Animal 10:262–273
Li Y, Kim J-J (2015) Effective population size and signatures of selection using bovine 50K SNP chips in Korean Native Cattle (Hanwoo). Evol Bioinform 11:EBO.S24359
Lu D, Sargolzaei M, Kelly M, Li C, Vander Voort G, Wang Z, Plastow G, Moore S, Miller SP (2012) Linkage disequilibrium in Angus, Charolais, and Crossbred beef cattle. Front Genetics 3:152
Makanjuola BO, Miglior F, Abdalla EA, Maltecca C, Schenkel FS, Baes CF (2020) Effect of genomic selection on rate of inbreeding and coancestry and effective population size of Holstein and Jersey cattle populations. J Dairy Sci 103:5183–5199
Makina S, Taylor J, van Marle-Kö ster E, Muchadeyi FC, Makgahlela ML, MacNeil MD et al (2015) Extent of linkage disequilibrium and effective population size in four South African Sanga cattle breeds. Front Genetics 6:337
Martinez R, Bejarano D, Ramírez J, Ocampo R, Polanco N, Perez JE, Onofre HG, Rocha JF (2023) Genomic variability and population structure of six Colombian cattle breeds. Trop Anim Health Prod 55:1–8
Matukumalli LK, Lawley CT, Schnabel RD, Taylor JF, Allan MF, Heaton MP, O’Connell J, Moore SS, Smith TP, Sonstegard TS (2009) Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 4:e5350
Mbole-Kariuki MN, Sonstegard T, Orth A, Thumbi S, Bronsvoort BDC, Kiara H, Toye P, Conradie I, Jennings A, Coetzer K (2014) Genome-wide analysis reveals the ancient and recent admixture history of East African Shorthorn Zebu from Western Kenya. Heredity 113:297–305
McKay SD, Schnabel RD, Murdoch BM, Matukumalli LK, Aerts J, Coppieters W, Crews D, Neto ED, Gill CA, Gao C (2007) Whole genome linkage disequilibrium maps in cattle. BMC Genet 8:1–12
Meuwissen TH, Hayes BJ, Goddard M (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Mrode R, Ojango JMK, Okeyo A, Mwacharo JM (2019) Genomic selection and use of molecular tools in breeding programs for indigenous and crossbred cattle in developing countries: current status and future prospects. Front Genet 9:694
Mumtaz S, Mukherjee A, Pathak P, Parveen K (2021) Effects of inbreeding on performance traits in Karan Fries crossbred cattle. Indian J Anim Sci 91(5):1
Murray C, Huerta-Sanchez E, Casey F, Bradley DG (2010) Cattle demographic history modelled from autosomal sequence variation. Philos Trans R Soc B Biol Sci 365:2531–2539
O’Brien AMP, Mészáros G, Utsunomiya YT, Sonstegard TS, Garcia JF, van Tassell CP, Carvalheiro R, da Silva MV, Sölkner J (2014) Linkage disequilibrium levels in Bos indicus and Bos taurus cattle using medium and high density SNP chip data and different minor allele frequency distributions. Livest Sci 166:121–132
Ouédraogo D, Ouédraogo-Koné S, Yougbaré B, Soudré A, Zoma-Traoré B, Mészáros G, Khayatzadeh N, Traoré A, Sanou M, Mwai OA (2021) Population structure, inbreeding and admixture in local cattle populations managed by community-based breeding programs in Burkina Faso. J Anim Breed Genet 138:379–388
Porto-Neto LR, Kijas JW, Reverter A (2014) The extent of linkage disequilibrium in beef cattle breeds using high-density SNP genotypes. Genet Sel Evol 46:1–5
Reich DE, Cargill M, Bolk S, Ireland J, Sabeti PC, Richter DJ, Lavery T, Kouyoumjian R, Farhadian SF, Ward R (2001) Linkage disequilibrium in the human genome. Nature 411:199–204
Rodríguez-Ramilo ST, Fernández J, Toro MA, Hernández D, Villanueva B (2015) Genome-wide estimates of coancestry, inbreeding and effective population size in the Spanish Holstein population. PLoS ONE 10:e0124157
Saravanan K, Panigrahi M, Kumar H, Parida S, Bhushan B, Gaur G, Kumar P, Dutt T, Mishra B, Singh R (2022) Genome-wide assessment of genetic diversity, linkage disequilibrium and haplotype block structure in Tharparkar cattle breed of India. Anim Biotechnol 33:297–311
Sargolzaei M, Schenkel F, Jansen G, Schaeffer L (2008) Extent of linkage disequilibrium in Holstein cattle in North America. J Dairy Sci 91:2106–2117
Silva C, Neves H, Queiroz S, Sena J, Pimentel E (2010) Extent of linkage disequilibrium in Brazilian Gyr dairy cattle based on genotypes of AI sires for dense SNP markers. In: Proceedings of the 9th world congress on genetics applied to livestock production: 1–6 August 2010. Leipzig, pp 1–29
Singh A, Kumar A, Mehrotra A, Pandey AK, Mishra B, Dutt T (2021) Estimation of linkage disequilibrium levels and allele frequency distribution in crossbred Vrindavani cattle using 50K SNP data. PLoS ONE 16:e0259572
Slifer SH (2018) PLINK: key functions for data analysis. Curr Protocols Human Genet 97:e59
Stachowicz K, Sargolzaei M, Miglior F, Schenkel F (2011) Rates of inbreeding and genetic diversity in Canadian Holstein and Jersey cattle. J Dairy Sci 94:5160–5175
Team RC (2020) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria
Tian R, Asadollahpour Nanaie H, Wang X, Dalai B, Zhao M, Wang F, Li H, Yang D, Zhang H, Li Y (2023) Genomic adaptation to extreme climate conditions in beef cattle as a consequence of cross-breeding program. BMC Genomics 24:1–10
Uimari P, Kontkanen O, Visscher PM, Pirskanen M, Fuentes R, Salonen JT (2005) Genome-wide linkage disequilibrium from 100,000 SNPs in the East Finland founder population. Twin Res Hum Genet 8:185–197
Villa-Angulo R, Matukumalli LK, Gill CA, Choi J, van Tassell CP, Grefenstette JJ (2009) High-resolution haplotype block structure in the cattle genome. BMC Genet 10:1–13
Wray NR (2005) Allele frequencies and the r2 measure of linkage disequilibrium: impact on design and interpretation of association studies. Twin Res Human Genet 8:87–94
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS ONE 4:e8451
Zhang H, Yin L, Wang M, Yuan X, Liu X (2019) Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations. Front Genet 10:189
Zhao H, Nettleton D, Dekkers JC (2007) Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between single nucleotide polymorphisms. Genetics Res 89:1–6
Zhao F, Wang G, Zeng T, Wei C, Zhang L, Wang H, Zhang S, Liu R, Liu Z, Du L (2014) Estimations of genomic linkage disequilibrium and effective population sizes in three sheep populations. Livest Sci 170:22–29
Zhu M, Zhu B, Wang Y, Wu Y, Xu L, Guo L, Yuan Z, Zhang L, Gao X, Gao H (2013) Linkage disequilibrium estimation of Chinese beef Simmental cattle using high-density SNP panels. Asian Aust J Anim Sci 26:772–779
Acknowledgements
We acknowledge the Higher Education Commission, Pakistan for providing funding to the first author for her Ph.D. Studies.
Funding
This study is funded by the Pakistan Agricultural Research Council, Agricultural Linkages Programme (ALP) with Project Identification No. AS 016 titled “Development and application of genomic selection in foreign and local cattle breeds for improvement in dairy-related traits”.
Author information
Authors and Affiliations
Contributions
Conceptualization: FUN, RM. Data curation: FUN, MA. Formal analysis: FUN, HK. Investigation: FUN. Methodology: FUN. Project administration: SM, ZM, IA. Resources: SM, IA. Software: FUN. Supervision: RM, ZM, SM. Visualization: FUN, HK. Writing-original draft: FUN. Writing-review and editing: RM, HK, ZM, MA, SM, IA.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
To ensure the ethical and humane treatment of animals, the study described in this research paper was approved by the Research Ethics Committee of the National Institute for Biotechnology and Genetic Engineering (NIBGE), Faisalabad, Pakistan on 10-06-2020. During blood collection, a professional veterinarian was there to ensure minimal distress and harm to the animals. Before collecting any samples, the researchers met with the owners of the farm where the animals were housed to explain the purpose of the study and obtain informed consent verbally.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nisa, F.u., Kaul, H., Asif, M. et al. Genetic insights into crossbred dairy cattle of Pakistan: exploring allele frequency, linkage disequilibrium, and effective population size at a genome-wide scale. Mamm Genome 34, 602–614 (2023). https://doi.org/10.1007/s00335-023-10019-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-023-10019-y