
Abstract
Primary hypoadrenocorticism, also known as Addison’s disease, is an autoimmune disorder leading to the destruction of the adrenal cortex and subsequent loss of glucocorticoid and mineralocorticoid hormones. The disease is prevalent in Standard Poodles and is believed to be highly heritable in the breed. Using genotypes derived from the Illumina Canine HD SNP array, we performed a genome-wide association study of 133 carefully phenotyped Standard Poodles (61 affected, 72 unaffected) and found no markers significantly associated with the disease. We also sequenced the entire genomes of 20 Standard Poodles (13 affected, 7 unaffected) and analyzed the data to identify common variants (including SNPs, indels, structural variants, and copy number variants) across affected dogs and variants segregating within a single pedigree of highly affected dogs. We identified several candidate genes that may be fixed in both Standard Poodles and a small population of dogs of related breeds. Further studies are required to confirm these findings more broadly, as well as additional gene-mapping efforts aimed at fully understanding the genetic basis of what is likely a complex inherited disorder.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Primary hypoadrenocorticism, or Addison’s disease (AD), is an immune-mediated disorder caused by an aberrant T cell response against the outer layers of the adrenal cortex (Hadlow 1953; Schaer et al. 1986; Mitchell and Pearce 2012; Frank et al. 2013; Boag et al. 2015; Cartwright et al. 2016). The disease is naturally occurring in both humans and dogs.
In humans, AD has a strong genetic component that influences disease onset and progression. Specific disease susceptibility alleles have been identified in genes including major histocompatibility complex type II genes, CTLA4, and PTPN22, among others (Mitchell and Pearce 2012). These alleles tend to confer moderately elevated odds ratios of developing AD (~1.3- to 3- fold), and most variants are found only in particular human sub-populations (Myhre et al. 2002; Blomhoff et al. 2004; Skinningsrud et al. 2008a, b). This genetic heterogeneity, as well as the well-documented incomplete penetrance of the identified polymorphisms, has led to the hypothesis that the disease in humans is a complex, polygenic disorder with environmental influences. A complete understanding of the genetics of AD in humans remains elusive.
In dogs, certain breeds are highly overrepresented with AD, including Standard Poodles, Portuguese Water Dogs (PWDs), Bearded Collies, and Nova Scotia Duck Tolling Retrievers (NSTDRs) (Hanson et al. 2015), strongly suggesting a genetic link. Pedigree studies have estimated a relatively high heritability in these breeds, ranging from 0.49 in PWDs to 0.98 in NSDTRs (Famula et al. 2003; Oberbauer et al. 2006; Hughes et al. 2007). Attempts to identify specific polymorphisms in dogs with AD have recently been reviewed (Boag and Catchpole 2014). All of these efforts have involved candidate gene studies and have identified some breed-specific polymorphisms with only modest disease associations.
Here, we describe the first reported genome-wide approach in dogs aimed at identifying polymorphisms associated with AD in Standard Poodles.Footnote 1 We focused on Standard Poodles because this breed has a relatively high incidence of the disease at ~8–10%, a high estimated heritability of 0.76, and a possible major causative locus with an autosomal recessive inheritance pattern (Famula et al. 2003). Based upon this information, while accounting for studies in humans suggesting a complex inheritance pattern, we hypothesized that a combination of a genome-wide association study (GWAS) and whole-genome sequencing (WGS) would allow us to identify specific causative variants that lead to the development of AD in Standard Poodles.
Methods
Animal selection/phenotyping
We collected pedigrees and whole blood samples from 77 Standard Poodles with AD and 103 without AD; we also collected blood samples from related breeds with AD including PWDs, Labradoodles, and Goldendoodles. Requirements for a diagnosis of AD were minimum age of 1 year, serum sodum:potassium ratio <27:1, ACTH stimulation test with pre- and post-ACTH cortisol levels <2 μg/dL, and a supportive clinical history (e.g., waxing/waning gastrointestinal upset, anorexia, weakness) (Van Lanen and Sande 2014). All unaffected dogs had to be at least 10 years of age and have either a baseline cortisol ≥2 μg/dL or a normal ACTH stimulation test (Lennon et al. 2007); we also verified that unaffected dogs had not been receiving any steroid-containing medications for at least one month prior to cortisol testing. Phenotypes were validated by a diplomate of the American College of Veterinary Emergency and Critical Care or the American College of Veterinary Internal Medicine. All blood was collected in EDTA tubes, and DNA was extracted using the standard protocol of the DNeasy Blood and Tissue Kit (Qiagen).
SNP array genotyping
Approximately 0.4 μg of DNA from all 180 Standard Poodles was submitted to Neogen/GeneSeek (Lincoln, NE) for processing and genotyping using the 174 K Canine HD BeadChip array (Illumina). Genotyping and variant calling were carried out using GenomeStudio (Illumina) per manufacturer recommendations.
Genotype imputation
Using a reference panel comprising ~4.9 million high-quality SNPs derived from whole-genome sequencing of 83 dogs across 15 breeds (including 20 Standard Poodles, see below), we imputed the genotypes of all dogs evaluated on the SNP array. Target panel pre-phasing was performed using SHAPEIT 2.2 (Delaneau et al. 2012) and imputation was performed using IMPUTE2 2.3.2 (Marchini et al. 2007; Howie et al. 2009). Complete details regarding reference and target panel development and imputation methodology are described elsewhere (Friedenberg and Meurs 2016).
Genome-wide association study
SNP array-derived genotypes and samples were filtered to exclude those dogs and SNPs meeting the following criteria: per-sample call rate <0.9, inferred gender inconsistency based upon chromosome X heterozygosity <0.05 for known males, per-SNP call rate <0.9, minor allele frequency <0.05, Hardy–Weinberg equilibrium p value <1 × 10−7 for control samples, or Mendelian genotype inconsistency based upon the available pedigree data. Principal components were then calculated, and sample outliers >1.5 × IQR based upon distance from the median centroid were removed using multi-dimensional scaling. The remaining SNPs and samples were used to calculate a kinship matrix based upon IBS distance, and a GWAS was performed with EMMAX (Kang et al. 2010) using the kinship matrix as a random effect to correct for cryptic population structure. A threshold of p = 5 × 10−7 was used to determine significance. All calculations were performed using Golden Helix SNP and Variation Suite 8.44 (Golden Helix, Bozeman, MT).
GWAS was also performed on genotypes derived from SNP imputation. Imputed genotype calls were first filtered to include only those with an IMPUTE2 Info parameter ≥0.7 in order to select those genotypes with the highest imputation accuracy. Identical filtering methods and GWAS methodology were used to evaluate the imputed genotype data.
Whole-genome sequencing
Approximately 3 μg of DNA from 20 Standard Poodles (13 affected, seven unaffected) was submitted for library preparation and whole-genome sequencing at the University of North Carolina Chapel Hill High-Throughput Sequencing Facility. All sequencing experiments were designed as 125-bp paired-end reads, and samples were run on either one or two lanes of an Illumina HiSeq 2500 high-throughput sequencing system.
Variant calling from WGS data was performed using a standardized bioinformatics pipeline for all samples as described previously (Friedenberg and Meurs 2016). We applied a variant quality score recalibration tranche sensitivity cutoff of 99.9% to SNPs and 99% to indels for use in downstream analyses; genotype calls with a phred-scaled quality score <20 were flagged but not removed from the variant callset.
Variant scoring
Variants were evaluated using Variant Effect Predictor (VEP) 83 (McLaren et al. 2010) with the addition of Gene Ontology (Ashburner et al. 2000), GERP++ (Davydov et al. 2010), and Gene Expression Atlas (Petryszak et al. 2016) plugins. Each variant was scored using a three-point system to account for the VEP-derived variant consequence, the GERP++ conservation score, and the predicted biologic function; scoring algorithm details are shown in Supplemental Fig. 1 (top). Scoring criteria were chosen in order to maximize the likelihood of capturing variants with an explainable effect on gene function and/or a role in adrenal gland or humoral immune system biology.
Analysis of whole-genome sequences
We analyzed whole-genome sequence data by polymorphism type including biallelic sites, multiallelic sites, structural variants, and copy number variants (CNVs). We also performed separate analysis of biallelic sites in a particular family of dogs with a high incidence of AD.
Biallelic sites
WGS variants were filtered for biallelic SNPs and indels, followed by a second filter to include only sites variant (heterozygous or homozygous) in at least 11/13 of the affected dogs. We chose this filter to select variants that are common among affected dogs (assuming that a major locus contributing to AD arose only once in Standard Poodles, but accounting for some genotyping errors) and to allow for low disease penetrance in unaffected dogs. We then removed those variants that were common among the 63 non-Poodles contained in our imputation reference panel using a MAF cutoff of ≥0.1 given the low incidence of AD among these 14 breeds (Hanson et al. 2015). The remaining variants were evaluated using the scoring criteria described above.
We flagged all three-point variants and certain two-point variants for follow-up Sanger sequencing in a population of six additional dogs with AD including two Standard Poodles (not among those submitted for WGS), two PWDs, one Labradoodle, and one Goldendoodle. Affected dogs from these additional breeds were chosen because they are highly related to Standard Poodles (vonHoldt et al. 2010), and we assumed that they might be likely to share AD-predisposing polymorphisms. Further details regarding the follow-up criteria for three- and two-point variants are shown in Supplemental Fig. 1 (bottom).
For polymorphisms that were consistent across Standard Poodles and the related breeds, we sequenced the variant in additional 36 dogs across 12 breeds (three dogs/breed) including Labrador Retriever, Newfoundland, Norfolk Terrier, English Springer Spaniel, Boston Terrier, Cavalier King Charles Spaniel, Pomeranian, English Bulldog, Chihuahua, Pug, Miniature Poodle, and mixed-breed dog. DNA samples from these dogs were acquired as a part of the ongoing disease-related research in our laboratory.
Multiallelic sites
Multiallelic sites were analyzed using similar methods to the biallelic sites. However, polymorphisms were not filtered at a site level using a single MAF cutoff from the 63 non-Poodles in the imputation reference panel. Instead, we used custom scripting in R to compare the alternate alleles in Standard Poodles to the alternate alleles in the 63 non-Poodles at each polymorphic site. We selected those sites where no alternate alleles matched between Poodles and non-Poodles, or where the MAF of a matching allele was ≤0.1 in the non-Poodles. This method allowed us to account for the differences in individual alleles and allele frequencies between the two populations at the multiallelic sites.
Structural variants
Structural variants including large deletions, large insertions, inversions, and tandem duplications were evaluated using Pindel (Ye et al. 2009). Variants were filtered to include polymorphic sites in a minimum of 11/13 affected dogs and at least 10 bp in length in order to minimize overlap with previously called GATK variants. Variant effects were evaluated using VEP, and sites meeting criteria for both variant consequence and biological function (Supplemental Fig. 1, top) were selected for follow-up (GERP++ conservation scores are not calculated for large structural variants). BAM and VCF files were then visually inspected at the structural variant sites to exclude those structural variants common in non-Poodles as Pindel was not run on the non-Poodle samples.
Copy number variants
We evaluated aligned BAM files for CNVs using cn.MOPS (Klambauer et al. 2012). We then reduced overlapping CNVs into contiguous ranges and counted CNV gains and losses within each range for each dog using the GenomicRanges package for R (Lawrence et al. 2013). Hits were totaled within each range by phenotype, and we searched for ranges in which at least 11/13 affected Standard Poodles had consistent copy number gains or copy number losses.
Pedigree-based analysis
We considered the possibility that multiple variants contributing to AD arose at different points in the development of the Standard Poodle breed. To investigate this possibility, we explored variants from whole-genome sequencing specific to a single family of Standard Poodles with a high prevalence of AD across four generations, assuming that a causative locus for AD might be specific to this family only (Fig. 1). WGS was performed on four dogs (D00870, D00871, D00868, D00920) and SNP array genotyping and imputation was performed on 11 dogs (all except D00950 and D01239; dogs whose IDs begin with a “U” were either unavailable or too young to accurately phenotype).

Pedigree drawing of one family of Standard Poodles with a high prevalence of Addison’s disease. Dogs shaded red are affected, dogs outlined in blue are unaffected, and dogs outlined in black were unavailable (died, not reachable, or too young to phenotype) (Color figure online)
Based upon inspection of the pedigree (roughly even distribution of affected dogs by gender, affected dogs arising from unaffected parents, increased disease frequency in consanguineous breedings), we assumed a possible autosomal recessive inheritance pattern for at least one AD-predisposing variant within this family (recognizing that a complex inheritance pattern cannot be excluded). Using GATK, we selected imputed sites in which genotype calls for dogs in this pedigree were as follows: homozygous variant for all affected dogs, heterozygous for D00136, D00873, D00920, and not homozygous variant for D00923. The resulting sites were scored using our three-point algorithm, and three- and two-point variants were sequenced to evaluate fit with an autosomal recessive pattern in all 13 pedigree dogs whose DNA was available.
Results
Samples
Among the 77 Standard Poodles with AD and 103 Standard Poodles without AD, 44 (57%) affected dogs were female and 62 (60%) unaffected dogs were female. Blood chemistry results used for phenotyping are summarized in Table 1. A subset of 20 of these dogs were submitted for whole-genome sequencing, including 13 affected dogs (six females) and seven unaffected dogs (four females).
Several (N = 39) of the dogs included in our study were known to be related. These included one family of 16 dogs (1 affected) spanning four generations, one family of 13 dogs (six affected) spanning four generations (Fig. 1), one family of four dogs (one affected) spanning two generations, one family of three dogs (two affected) spanning three generations, and one family of three dogs (one affected) spanning two generations. All of these dogs were genotyped using the SNP array; four dogs (three affected) from the family of 13 dogs and three dogs (one affected) from the family of 16 dogs were among those submitted for whole-genome sequencing. We removed related dogs in certain analyses (e.g., GWAS) but took advantage of known relationships in others (e.g., pedigree analysis).
GWAS
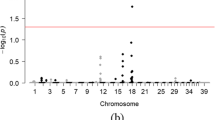
After initial filtering, 175 samples and 137,714 SNPs remained. Four samples (two affected, two unaffected) had a call rate <0.9, and one sample (affected) was flagged for gender inconsistency; among the 35,968 SNPs that were filtered, 32,620 had a MAF <0.05; 3496 had a call rate <0.9; 3109 had Mendelian inconsistencies; and 391 had a HWE p-value <1 × 10−7 for control samples. Principal component plots for the first and second eigenvalues using filtered SNPs and samples are shown in Supplemental Fig. 2. Based upon MDS outlier analysis, 42 dogs (13 affected, 29 unaffected, including all dogs known to be related) were removed from downstream GWAS analysis (Supplemental Fig. 2, Panel A). A case-control GWAS based upon the remaining 133 samples (61 affected, 72 unaffected) showed no SNPs or loci meeting genome-wide significance for association with AD in Standard Poodles (Fig. 2). A case-control GWAS using 2,641,537 SNPs derived from genotype imputation and the same 133 samples also failed to show any loci meeting genome-wide significance.

Summary of GWAS findings. Manhattan plot (a) and QQ plot (b) of case-control genome-wide association test performed using 61 cases and 72 controls of Standard Poodles with and without Addison’s disease. No SNPs met the genome-wide threshold for a significance of p = 5 × 10−7 (dashed red line, a) (Color figure online)
Whole-genome sequencing
Average coverage for whole-genome sequences ranged from 21× to 47×. After variant calling and filtering, 11,697,324 variants remained. Of these variants, 8,661,177 were biallelic SNPs; 2,468,209 were biallelic indels; 18,805 were multiallelic SNPs; and 463,350 were multiallelic indels.
Biallelic sites
We identified 2,531,187 biallelic sites (SNPs and indels) polymorphic in at least 11/13 Standard Poodles with AD. Of these variants, 64,745 had a MAF <0.1 in non-Poodle dogs and 421 had a score of at least two points (Fig. 3). The 19 variants with three points were found in six genes: CCR7, NR1D1, RXRA, FLT3, HNRNPA1, and PTPN6. The 402 variants with two points were found in 192 genes. After curating these 402 variants (see Supplemental Fig. 1, bottom, for details), we selected polymorphisms in 26 of these genes for additional sequencing: SKAP1, MED1, CH25H, ZNF385C, CD79B, PSMC1, NR2E1, SRD5A3, DLG1, GIMAP8, CCDC88C, CYP1A2, MED24, PSMD3, SEL1L3, UBASH3A, UNC119, RARG, LEXM, RIPK2, ITSN2, THRA, CCR7, WDR46, CD69, and PMAIP1.

Venn diagram of biallelic SNPs and indels derived from whole-genome sequencing common to 11/13 Standard Poodles with Addison’s disease. Variants were filtered to remove common sites (MAF ≥ 0.1) among a population of 63 non-Poodles across 14 additional dog breeds and subsequently scored as described in Supplemental Fig. 1
We sequenced 34 variants in these genes using a validation cohort of six dogs (two Standard Poodles, two PWDs, one Goldendoodle, and one Labradoodle) with AD. None of these variants were consistently present across the validation cohort (Supplemental Table 1). Two polymorphisms in PMAIP1 and PSMC1 were present in the Standard Poodles, Goldendoodle, and Labradoodle, but not the PWDs. Nineteen variants were present in Standard Poodles only; four of these had three points using our scoring algorithm (in CCR7 [two variants], NR1D1, and RXRA). Thirteen variants were found sporadically across the validation cohort. We genotyped the six variants in PMAIP1, PSMC1, CCR7, NR1D1, and RXRA in 36 additional non-Poodles (12 breeds), and found all of these variants to be relatively uncommon in these breeds. For the variants present in affected Standard Poodles, the Goldendoodle, and the Labradoodle, the downstream gene variant in PMAIP1 was present only in one Boston Terrier, and the 3′-UTR variant in PSMC1 was present in one Cavalier King Charles Spaniel, one Pomeranian, and one Pug (Supplemental Table 1). For the variants common across affected Standard Poodles in our validation cohort, the 3′-UTR variants in CCR7 were present in one Pomeranian; the missense variant in NR1D1 was present in one mixed-breed dog and one English Springer Spaniel; and the 3′-UTR variant in RXRA was present in one Chihuahua and one Pug (Supplemental Table 1).
Multiallelic sites
We identified 166,615 multiallelic sites (SNPs and indels) polymorphic in at least 11/13 Standard Poodles with AD. Of these variants, 1917 met our site-level filtering criteria based upon the absence of common alleles between Poodle and non-Poodle dogs. After scoring these variants using VEP, one variant had three points and seven variants had two points. All of these multiallelic sites were located in repetitive regions of the genome where next-generation sequencing is challenging and genotype calls are often unreliable (Treangen and Salzberg 2012). We therefore visually inspected all of the three- and two-point variants, and determined that none of the multiallelic sites were strong candidates for follow-up Sanger sequencing in our validation cohort of non-Poodle dogs as there was no consistency or pattern among the genotypes of the affected dogs.
Structural variants
We identified 168,706 structural variants common to 11/13 Standard Poodles using Pindel; 17,093 remained after removing those sites less than 10 bp in length (Supplemental Table 2). After further subsetting these polymorphisms to include those meeting scoring criteria for both variant consequence and biological function, 11 deletions and four small insertions remained. All of these variants had been previously identified by GATK and were common among our population of 63 non-Poodle dogs, and were therefore not considered for follow-up Sanger sequencing in any additional dogs.
Copy number variants
We identified 3202 CNVs across the genome, with a median length of 6 Kb (range 3 Kb to 6.53 Mb) (Supplemental Table 3). The largest CNV represents the pseudo-autosomal region at the 5′ end of the X chromosome. We did not, however, identify any CNV gains or losses that were consistent across at least 11/13 affected Standard Poodles.
Pedigree-based analysis
We identified 4539 variants (SNPs and indels) consistent with an autosomal recessive inheritance pattern using imputed genotypes in the 11 related dogs described in Fig. 1. Of these variants, one had a score of three points and seven variants in five genes had a score of two points. We selected two polymorphisms in TNIP3 and one polymorphism in IL4R and PCSK5 for follow-up Sanger sequencing in the entire pedigree based upon their known biological function. However, none of these variants were consistent with an autosomal recessive inheritance pattern when all 13 pedigree dogs were sequenced (Supplemental Table 4).
Discussion
In this study, we used several methods to identify genetic polymorphisms that cause or contribute to the development of AD in Standard Poodles. Based upon our analysis, we were unable to pinpoint any specific causative variants. However, our findings suggest some possibilities regarding the genetic architecture of the disease, as well as important avenues for further investigation.
After sample filtering, our GWAS contained 61 affected and 72 unaffected dogs. All samples were stringently phenotyped for either the presence or absence of disease. Based upon prior case-control studies in dogs (Kyöstilä et al. 2012; Tengvall et al. 2013; Safra et al. 2013; Jagannathan et al. 2013; Wolf et al. 2014; Drögemüller et al. 2014), as well as power analyses using the Illumina 174 K HD SNP array (Lequarré et al. 2011), this sample size should have been able to detect a large-effect locus that distinguishes affected from unaffected dogs within the breed. Our findings, as well as the unpublished reports of others (Footnote 1), suggest that such a locus is unlikely to exist and that additional strategies are required to explain the genetic underpinnings and high prevalence of the disease within Standard Poodles.
One recent study examining the effects of genetic bottlenecks within the breed is consistent with our GWAS findings (Pedersen et al. 2015). By genotyping 33 highly discriminatory microsatellite markers in samples acquired across North America and Europe, the study authors determined that Standard Poodle dogs with AD tend to be highly inbred. Furthermore, based upon their analysis of internal relatedness and heterozygosity among these inbred dogs, the authors suggest that the loci responsible for AD may be fixed in a majority of both affected and unaffected Standard Poodles. This conclusion would support our inability to detect a single causative locus for AD in Standard Poodles using a GWAS of only 133 dogs.
Based upon our negative GWAS, we analyzed WGS data from 13 affected and seven unaffected dogs. We looked for disease-relevant polymorphisms, structural variants, and CNVs. In these analyses, we ignored the unaffected dog population given our GWAS findings which failed to show any markers that distinguish these populations, as well as the hypothesis that autoimmune diseases may have strong environmental influences leading to incomplete disease penetrance (Bogdanos et al. 2013; Colafrancesco et al. 2013).
In analyzing our WGS data without the assistance of a statistically significant marker, we were limited to a priori assumptions regarding the pathogenesis of the disease. We searched for variants that could be relevant to either humoral immunity or the adrenal gland, coupled with an understanding of transcript structure based upon the existing gene annotations and GERP++ conservation scores. While we identified some candidate polymorphisms in this analysis (Supplemental Table 1), none of the variants we evaluated in a larger population of dogs were common across all related breeds in our validation cohort nor completely absent in a population of unrelated dog breeds. This might suggest that our prioritization of AD-predisposing variants is incorrect and that additional genotyping of lower-priority variants in a larger population of affected Standard Poodles and additional unrelated breeds is indicated. However, it could equally suggest that our analysis of WGS data was limited by our a priori (but unavoidable) assumptions regarding gene function. Indeed, many disease-causing mutations in dogs have been identified in genes that might at first glance seem to be unlikely candidates (Wiik et al. 2008; Meurs et al. 2010; Downs et al. 2011), reinforcing the benefit of successful association mapping in interpreting genome-wide data. Additionally, our WGS analysis was constrained by the number of dogs we were able to sequence, thereby limiting our ability to apply statistical association testing to the WGS data directly.
We also analyzed our WGS data by tracing polymorphisms through a pedigree of dogs with a high prevalence of AD (Fig. 1). Here too, we identified some candidate genes using imputed genotypes; however, upon further sequencing none of these variants remained consistent with the autosomal recessive inheritance pattern suggested by the pedigree. We prioritized variants for follow-up sequencing using existing gene annotations, which again could explain our failure to identify an AD-predisposing locus within the family. Another possibility, however, might be that even within this pedigree, AD has a complex inheritance pattern that is not easily discernable given multi-locus effects or gene–gene interactions.
Beyond the approaches we attempted here, there are several other reasonable gene-mapping strategies that could help identify causative or predisposing variants for AD in Standard Poodles going forward. One important approach could be a cross-breed GWAS using a denser array of markers as described recently for several other complex diseases in dogs (Hayward et al. 2016). Such an approach could flag associations shared among dogs with AD of different ancestries, but would require a significantly larger sample set (~500–1000 cases and controls). Alternatively, whole-genome sequencing of affected cross-breed dogs such as Labradoodles or Goldendoodles could help narrow the search for disease-associated variants and minimize the extent to which WGS relies on existing gene annotations. Lastly, gene expression studies of affected adrenocortical tissue could be helpful as well, but these are likely to be limited by challenges associated with sample collection.
In summary, our analysis of AD in Standard Poodles did not identify a single, major locus associated with the disease. This finding suggests that AD may either be a complex trait or relatively fixed within the Standard Poodle breed. Further genotyping studies that rely on a larger population of dogs or employ a cross-breed approach could be helpful in identifying disease-associated loci and/or causative polymorphisms going forward.
Notes
We are aware of one unpublished report of a genome-wide association study of 120 affected and 120 unaffected Standard Poodles performed at the Broad Institute that failed to identify any genetic differences between these two groups (The Broad Institute).
References
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. doi:10.1038/75556
Blomhoff A, Lie BA, Myhre AG et al (2004) Polymorphisms in the cytotoxic T lymphocyte antigen-4 gene region confer susceptibility to Addison’s disease. J Clin Endocrinol Metab 89:3474–3476. doi:10.1210/jc.2003-031854
Boag AM, Catchpole B (2014) A review of the genetics of hypoadrenocorticism. Top Companion Anim Med 29:96–101. doi:10.1053/j.tcam.2015.01.001
Boag AM, Christie MR, McLaughlin KA et al (2015) Autoantibodies against cytochrome P450 side-chain cleavage enzyme in dogs (Canis lupus familiaris) affected with hypoadrenocorticism (Addison’s Disease). PLoS ONE 10:e0143458. doi:10.1371/journal.pone.0143458
Bogdanos DP, Smyk DS, Invernizzi P et al (2013) Tracing environmental markers of autoimmunity: introducing the infectome. Immunol Res 56:220–240. doi:10.1007/s12026-013-8399-6
Cartwright JA, Stone J, Rick M, Dunning MD (2016) Polyglandular endocrinopathy type II (Schmidt’s syndrome) in a Dobermann pinscher. J Small Anim Pract 57:491–494. doi:10.1111/jsap.12535
Colafrancesco S, Agmon-Levin N, Perricone C, Shoenfeld Y (2013) Unraveling the soul of autoimmune diseases: pathogenesis, diagnosis and treatment adding dowels to the puzzle. Immunol Res 56:200–205. doi:10.1007/s12026-013-8429-4
Davydov EV, Goode DL, Sirota M et al (2010) Identifying a high fraction of the human genome to be under selective constraint using GERP++. PLoS Comp Biol 6:e1001025. doi:10.1371/journal.pcbi.1001025
Delaneau O, Marchini J, Zagury J-F (2012) A linear complexity phasing method for thousands of genomes. Nat Methods 9:179–181. doi:10.1038/nmeth.1785
Downs LM, Wallin-Håkansson B, Boursnell M et al (2011) A frameshift mutation in golden retriever dogs with progressive retinal atrophy endorses SLC4A3 as a candidate gene for human retinal degenerations. PLoS ONE 6:e21452. doi:10.1371/journal.pone.0021452
Drögemüller M, Jagannathan V, Becker D et al (2014) A Mutation in the FAM83G gene in dogs with Hereditary footpad hyperkeratosis (HFH). PLoS Genet 10:e1004370. doi:10.1371/journal.pgen.1004370
Famula TR, Belanger JM, Oberbauer AM (2003) Heritability and complex segregation analysis of hypoadrenocorticism in the standard poodle. J Small Anim Pract 44:8–12
Frank CB, Valentin SY, Scott-Moncrieff JCR, Miller MA (2013) Correlation of inflammation with adrenocortical atrophy in canine adrenalitis. J Comp Pathol 149:268–279. doi:10.1016/j.jcpa.2012.11.242
Friedenberg SG, Meurs KM (2016) Genotype imputation in the domestic dog. Mamm Genome. doi:10.1007/s00335-016-9636-9
Friedenberg SG, Meurs KM, Mackay TFC (2016) Evaluation of artificial selection in Standard Poodles using whole-genome sequencing. Mamm Genome. doi:10.1007/s00335-016-9660-9
Hadlow WJ (1953) Adrenal cortical atrophy in the dog; report of three cases. Am J Path 29:353–361
Hanson JM, Tengvall K, Bonnett BN, Hedhammar A (2015) Naturally occurring adrenocortical insufficiency—an epidemiological study based on a Swedish-insured dog population of 525,028 dogs. J Vet Intern Med. doi:10.1111/jvim.13815
Hayward JJ, Castelhano MG, Oliveira KC et al (2016) Complex disease and phenotype mapping in the domestic dog. Nat Commun 7:10460. doi:10.1038/ncomms10460
Howie BN, Donnelly P, Marchini J (2009) A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5:e1000529. doi:10.1371/journal.pgen.1000529
Hughes AM, Nelson RW, Famula TR, Bannasch DL (2007) Clinical features and heritability of hypoadrenocorticism in Nova Scotia Duck Tolling Retrievers: 25 cases (1994–2006). J Am Vet Med Assoc 231:407–412. doi:10.2460/javma.231.3.407
Jagannathan V, Bannoehr J, Plattet P et al (2013) A mutation in the SUV39H2 gene in Labrador Retrievers with hereditary nasal parakeratosis (HNPK) provides insights into the epigenetics of keratinocyte differentiation. PLoS Genet 9:e1003848. doi:10.1371/journal.pgen.1003848
Kang HM, Sul JH, Service SK et al (2010) Variance component model to account for sample structure in genome-wide association studies. Nat Genet 42:348–354
Klambauer G, Schwarzbauer K, Mayr A et al (2012) cn.MOPS: mixture of poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res 40:e69–e69. doi:10.1093/nar/gks003
Kyöstilä K, Cizinauskas S, Seppälä EH et al (2012) A SEL1L mutation links a canine progressive early-onset cerebellar ataxia to the endoplasmic reticulum-associated protein degradation (ERAD) machinery. PLoS Genet 8:e1002759. doi:10.1371/journal.pgen.1002759
Lawrence M, Huber W, Pagès H et al (2013) Software for computing and annotating genomic ranges. PLoS Comp Biol 9:e1003118. doi:10.1371/journal.pcbi.1003118
Lennon EM, Boyle TE, Hutchins RG et al (2007) Use of basal serum or plasma cortisol concentrations to rule out a diagnosis of hypoadrenocorticism in dogs: 123 cases (2000-2005). J Am Vet Med Assoc 231:413–416. doi:10.2460/javma.231.3.413
Lequarré A-S, Andersson L, André C et al (2011) LUPA: a European initiative taking advantage of the canine genome architecture for unravelling complex disorders in both human and dogs. Vet J 189:155–159. doi:10.1016/j.tvjl.2011.06.013
Marchini J, Howie B, Myers S et al (2007) A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 39:906–913. doi:10.1038/ng2088
McLaren W, Pritchard B, Rios D et al (2010) Deriving the consequences of genomic variants with the ensembl API and SNP effect predictor. Bioinformatics 26:2069–2070. doi:10.1093/bioinformatics/btq330
Meurs KM, Mauceli E, Lahmers S et al (2010) Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet 128:315–324. doi:10.1007/s00439-010-0855-y
Mitchell AL, Pearce SHS (2012) Autoimmune Addison disease: pathophysiology and genetic complexity. Nat Rev Endocrinol 8:306–316. doi:10.1038/nrendo.2011.245
Myhre AG, Undlien DE, Løvås K et al (2002) Autoimmune adrenocortical failure in Norway autoantibodies and human leukocyte antigen class II associations related to clinical features. J Clin Endocrinol Metab 87:618–623. doi:10.1210/jcem.87.2.8192
Oberbauer AM, Bell JS, Belanger JM, Famula TR (2006) Genetic evaluation of Addison’s disease in the Portuguese water dog. BMC Vet Res 2:15. doi:10.1186/1746-6148-2-15
Pedersen NC, Brucker L, Tessier NG et al (2015) The effect of genetic bottlenecks and inbreeding on the incidence of two major autoimmune diseases in standard poodles, sebaceous adenitis and Addison’s disease. Canine Genet Epidemiol 2:14. doi:10.1186/s40575-015-0026-5
Petryszak R, Keays M, Tang YA et al (2016) Expression Atlas update–an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res 44:D746–D752. doi:10.1093/nar/gkv1045
Safra N, Bassuk AG, Ferguson PJ et al (2013) Genome-wide association mapping in dogs enables identification of the homeobox gene, NKX2-8, as a genetic component of neural tube defects in humans. PLoS Genet 9:e1003646. doi:10.1371/journal.pgen.1003646
Schaer M, Riley WJ, Buergelt CD et al (1986) Autoimmunity and Addison’s disease in the dog. J Am Anim Hosp Assoc 22:786–794
Skinningsrud B, Husebye ES, Gervin K et al (2008a) Mutation screening of PTPN22: association of the 1858T-allele with Addison’s disease. Eur J Hum Genet 16:977–982. doi:10.1038/ejhg.2008.33
Skinningsrud B, Husebye ES, Pearce SH et al (2008b) Polymorphisms in CLEC16A and CIITA at 16p13 are associated with primary adrenal insufficiency. J Clin Endocrinol Metab 93:3310–3317. doi:10.1210/jc.2008-0821
Tengvall K, Kierczak M, Bergvall K et al (2013) Genome-wide analysis in German shepherd dogs reveals association of a locus on CFA 27 with atopic dermatitis. PLoS Genet 9:e1003475. doi:10.1371/journal.pgen.1003475
The Broad Institute Addison’s disease. https://www.broadinstitute.org/scientific-community/science/projects/mammals-models/dog/disease-research/addison%E2%80%99s-disease. Accessed 26 May 2016
Treangen TJ, Salzberg SL (2012) Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13:36–46. doi:10.1038/nrg3117
Van Lanen K, Sande A (2014) Canine hypoadrenocorticism: pathogenesis, diagnosis, and treatment. Top Companion Anim Med 29:88–95. doi:10.1053/j.tcam.2014.10.001
vonHoldt BM, Pollinger JP, Lohmueller KE et al (2010) Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464:898–902. doi:10.1038/nature08837
Wiik AC, Wade C, Biagi T et al (2008) A deletion in nephronophthisis 4 (NPHP4) is associated with recessive cone-rod dystrophy in standard wire-haired dachshund. Genome Res 18:1415–1421. doi:10.1101/gr.074302.107
Wolf ZT, Leslie EJ, Arzi B et al (2014) A LINE-1 insertion in DLX6 is responsible for cleft palate and mandibular abnormalities in a canine model of Pierre Robin sequence. PLoS Genet 10:e1004257. doi:10.1371/journal.pgen.1004257
Ye K, Schulz MH, Long Q et al (2009) Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25:2865–2871. doi:10.1093/bioinformatics/btp394
Acknowledgements
SGF is supported by a National Institutes of Health T32 training award (5T32OD011130-07). Funding for whole-genome sequencing was provided in part by the Poodle Club of America Foundation and the Morris Animal Foundation. Seed funding for this project was provided by the North Carolina State University Comparative Medicine Institute. Some whole-genome sequencing data were graciously contributed by Drs. Leigh Anne-Clark (13 dogs), Natasha J. Olby and Theirry Olivry (11 dogs), and Joshua A. Stern (two dogs).
Author contributions
SGF collected samples, designed the study, analyzed the data, and wrote the manuscript. KFL collected samples and provided guidance regarding phenotyping of dogs. KMM collected samples and supervised the study. All authors have read and edited the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
335_2016_9671_MOESM1_ESM.tiff
Supplemental figure 1—Scoring algorithm and follow-up criterial used to evaluate genetic variants for Addison’s disease. Each variant was allocated one point for meeting criteria in each of three categories: variant consequence, GERP++ conservation score, or biological function (top). Scoring was carried out using custom scripting/filtering in R. A maximum of 3 points was allocated to each variant. After each variant was scored, specific follow-up criteria were applied by manual curation (bottom). Variants flagged for follow-up were evaluated in additional population of dogs as described in the Methods section of the text. See http://useast.ensembl.org/info/genome/variation/predicted_data.html#consequences for a complete listing of VEP variant consequences. (TIFF 1848 kb)
335_2016_9671_MOESM2_ESM.tiff
Supplemental figure 2—Principal components plots of the first and second eigenvalues for 175 Standard Poodles passing initial filtering criteria. Individual dogs are colored to indicate (A) MDS outliers, (B) phenotype, and (C) gender. In (A), gray squares represent the 42 dogs that were removed from from downstream GWAS analysis based upon MDS outlier detection. (TIFF 36918 kb)
Rights and permissions
About this article

Cite this article
Friedenberg, S.G., Lunn, K.F. & Meurs, K.M. Evaluation of the genetic basis of primary hypoadrenocorticism in Standard Poodles using SNP array genotyping and whole-genome sequencing. Mamm Genome 28, 56–65 (2017). https://doi.org/10.1007/s00335-016-9671-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-016-9671-6