
Abstract
Meat quality traits have economically significant impacts on the pig industry, and can be improved using molecular approaches in pig breeding. Since 1994 when the first genome-wide scan for quantitative trait loci (QTLs) in pig was reported, over the past two decades, numerous QTLs have been identified for meat quality traits by family based linkage analyses. However, little is known about the genetic variants for meat quality traits in Chinese purebred or outbred populations. To unveil it, we performed a genome-wide association study for 10 meat quality traits in Chinese purebred Laiwu pigs. In total, 75 significant SNPs (P < 1.01 × 10−6) and 33 suggestive SNPs (P < 2.03 × 10−5) were identified. On SSC12, a region between 56.22 and 61.49 Mb harbored a cluster of SNPs that were associated with meat color parameters (L*, lightness; a*, redness; b*, yellowness) and moisture content of longissimus muscle (LM) and semimembranosus muscle at the genome-wide significance level. A region on SSC4 also has pleiotropic effects on moisture content and drip loss of LM. In addition, this study revealed at least five novel QTLs and several candidate genes including 4-linked MYH genes (MYH1, MYH2, MYH3, and MYH13), MAL2, LPAR1, and PRKAG3 at four significant loci. Except for the SSC12 QTL, other QTLs are likely tissue-specific. These results provide new insights into the genetic basis of meat quality traits in Chinese Laiwu pigs and some significant SNPs reported here could be incorporated into the selection programs involving this breed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Pork is a major meat resource for human. Pork quality has a great influence on consumer preference (Nonneman et al. 2013). So improving pork quality is a high priority in the pork industry (Becker et al. 2013). The major meat quality traits of interest, including pH, drip loss, meat color, moisture, and intramuscular fat contents, are common selection objectives in pig breeding programs (Rosenvold and Andersen 2003; Uimari and Sironen 2014). However, most of these traits show a low to moderate heritability and are difficult and expensive to measure (Suzuki et al. 2005). Their improvement by target breeding programs is challenging and requires knowledge of the genetic and molecular background (Becker et al. 2013).
Since the first genome-wide scan for quantitative trait loci (QTLs) in pigs was reported (Andersson et al. 1994), extensive studies have been conducted to identify QTLs in many experimental crosses. To date, 10,497 QTLs for 649 different traits have been deposited in the Pig QTL database (http://www.animalgenome.org/cgi-bin/QTLdb/SS/index). However, most of these QTLs were previously detected by linkage analyses of pig families with low-density microsatellite markers, which lead to lack of precision of their locations (Sanchez et al. 2006; Riquet et al. 2011). As a result, only a handful of genes, including RYR1, PRKAG3, IGF2, MCR4, and PHKG1, have so far been shown to affect meat quality and production traits in pig (Fujii et al. 1991; Kim et al. 2000; Milan et al. 2000; Van Laere et al. 2003; Ma et al. 2014). Moreover, QTL linkage designs have in most cases based on crosses between divergent populations (Rothschild and Bidanel 2002), resulting in QTL data that are not directly transferable to commercial populations (Sanchez et al. 2014). Compared to linkage mapping method, genome-wide association studies (GWAS) using high-density molecular markers can map QTLs at high resolution and pinpoint the most significantly associated markers for various traits in human and animals. Since the illumina porcine 60 K SNP Beadchip array is available (Ramos et al. 2009), some researchers have recently performed GWAS to detect QTLs in different crossbred and outbred pig populations, particularly in several western commercial pig lines (Ren et al. 2011; Luo et al. 2012; Ma et al. 2013; Ponsuksili et al. 2014; Sanchez et al. 2014; Stratz et al. 2014; Zhang et al. 2014). However, few studies have dissected QTLs for meat quality traits in Chinese indigenous breeds.
The Laiwu breed is the North China type pig representative, with tender meat, bright color, excellent flavor, high water holding capacity, and intramuscular fat content; and it has enjoyed a good reputation for long time. In this work, we carried out a GWAS of meat quality traits which were recorded on two different muscles, i.e., M. longissimus (LM) and M. semimembranosus (SM) muscles in 316 Laiwu pigs. This study aims to reveal genetic loci associated with meat quality traits and discover appealing candidate genes for follow-up investigations of the underlying genes and variants.
Materials and methods
Ethics statement
All procedures involving animals followed the guidelines for the care and use of experimental animals approved by the State Council of the People’s Republic of China. The ethics committee of Jiangxi Agriculture University specially approved this study.
Experimental population
The Laiwu population used in this study comprised 316 offspring produced by 12 boars and 45 sows. They were born and raised for 2–3 months on Laiwu Pig Breeding Center in Laiwu city, Shandong Province. They were then transferred to a farm in Nanchang city in four batches (Sep. 2, Nov. 2, 2012 and Jan. 24, May 10, 2013). All the males were castrated and all piglets were weaned at age of 21 or 22 days. They were fed on a similar diet under a standardized feeding and management regimen, and given free access to water. At the age of 300 days, a total of 316 individuals including 218 barrows and 98 gilts were slaughtered at a commercial abattoir in Nanchang.
Phenotype measurement
The longissimus muscle (LM) between the 10th-rib and the first lumbar vertebra and the semimembranosus muscle (SM) were evaluated for meat quality traits (Duan et al. 2009; Ma et al. 2009). Here, we analyzed the data of the meat quality traits including pH, drip loss, L* for lightness, a* for redness, b* for yellowness, and moisture content. Muscle pH was measured at 45 min and 24 h postmortem in LM and SM using a Delta 320 pH Meter. Each sample was measured twice and the average of parallel measurements was then used for subsequent analysis. Muscle color measurements were taken on the exposed cut surface of the two muscles with a CM-2600d/2500d Minolta Chroma Meter at 24 h postmortem. Color scores were subjectively assessed according to the color standard (1 = pale; 6 = dark) provided by US National Pork Procedures Council (NPPC) (Berg 2006). Drip loss after 24 h storage of LM and SM was measured using an EZ-DripLoss method (Rassmussen and Andersson 1996). Moisture content was determined by the routine oven-drying method. In this study, 316 Laiwu pigs were phenotyped. We also determined heritability estimates of 15 meat quality traits. Most of the meat quality traits show low to moderate heritabilities. Summary statistics for the 15 traits are in Table 1.
Genotyping and quality control
Genomic DNA was isolated from ear tissue of each sample using a standard phenol/chloroform method. The quality and concentration of DNA were determined by Nanodrop-1000 spectrophotometer (Thermo Fisher, USA). All DNA samples were qualified and standardized into a final concentration of 50 ng/ul.
A total of 316 animals were genotyped for 61,565 SNPs using the illumina porcine 60 K SNP beadchip according to the manufacture’s protocol. Quality control was carried out using Plink v1.07 (Purcell et al. 2007). We discarded SNPs with a call rate greater than 90 %, a minor allele frequency (MAF) smaller than 0.01 and a significant deviation from Hardy–Weinberg equilibrium (P ≤ 10−5). In addition, we removed animals with call rate less than 90 %. After filtering, a final set of 49452 SNPs for 316 Laiwu pigs were included for further statistical analysis. We used BLASTN to determine the positions of the SNPs in the Sscrofa 10.2 genome assembly.
Statistical analyses
The differences in meat quality traits between LM and SM in Laiwu pig were analyzed using the anov function of R software. Heritability of meat quality trait was estimated using the polygenic function of GenABEL v1.7 (Aulchenko et al. 2007). The GWAS was performed by polygenic followed by mmscore function of GenABEL v1.7 (Breslow and Clayton 1993; Yu et al. 2006; Bradbury et al. 2007). A generalized linear mixed model was used to estimate associations between SNPs and phenotypic values. Sex and Batch were fitted as fixed effects in the model. The significance thresholds were determined by Bonferroni method, in which the conventional P value was divided by the number of tests performed (Yang et al. 2005). A SNP was considered to have the stringent genome-wide significance at P < 0.05/N, and have the suggestive significance at P < 1/N, where N stands for the number of SNPs in the date analyses. In this study, the genome-wide and suggestive significance thresholds were 1.01e−06 (0.05/49452) and 2.02e−05 (1/49452), respectively. The phenotypic variance explained by the peak SNP was estimated by (Vreduce−Vfull)/Vreduce, where Vfull and Vreduce are residual variances of models for association analysis with and without SNP term, respectively.
The problem of population stratification was assessed by examining the distribution of test statistics generated from the thousands of association tests and assessing their deviation from the null distribution (i.e., the distribution expected under the null hypothesis of no SNP associated with the trait) in a quantile–quantile (Q–Q) plot (Pearson and Manolio 2008). In the Q–Q plot, the horizontal axis represents the expected-log10 P and the vertical axis represents the observed −log10 P. The diagonal line represents y = x, the shaded region shows 95 % confidence interval based on Beta distribution. Overall deviation above the diagonal identity line in the initial stage may suggest a severe population stratification. Deviations from the diagonal line suggest that either the assumed distribution is incorrect or that the sample contains values arising in some other manner, as by a true association (Wellcome Trust Case Control 2007). The Q–Q plot was generated using R software.
Haplotype or linkage disequilibrium (LD) block analyses were performed for the chromosomal regions with multiple significant SNPs clustered around the peak SNP. The LD blocks were determined using Haploview version 4.2 software with default settings (Barrett et al. 2005). A haplotype association study (Druet and Farnir 2011) was also performed to identify genomic regions associated with the LM_MoistureContent, LM_ColorM_L24 h, and LM_ColorM_a24 h. Haplotypes corresponding to a predetermined number (K = 20) of hidden haplotype states (Sartelet et al. 2012) were conducted with a hidden Markov model via PHASEBOOK (Druet and Georges 2010).
Results
Effects of muscle tissues on meat quality traits
Meat quality depends on skeletal muscle structure and metabolic properties (Herault et al. 2014). Although LM and SM are both classified as glycolytic muscles, they exhibit dissimilar myofiber composition and metabolic characteristics. Here, we compared muscle characteristics between LM and SM in the Laiwu pigs. Very significant (P < 0.001) differences were found between them in all seven characteristics (Table 2).
Assessment of population stratification
Population stratification is recognized as a major threat to the validity of GWAS results (Pearson and Manolio 2008). The Q–Q plots of test statistics of GWAS are shown in Supplementary Fig. S1. In this study, the average genomic inflation factors (λ) of the GWAS for all meat quality traits were 1.08, which suggests that there were no obvious population stratifications.
GWAS analyses
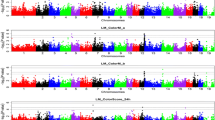
The GWAS results are illustrated in Fig. 1, Supplementary Fig. S2 and Table 3. A total of 108 SNPs passing the suggestive significance level were identified on six pig chromosomes (SSC) for 10 meat quality traits. Of the 108 SNPs, 75 SNPs and 33 SNPs reached the genome-wide and suggestive significance levels, respectively.

Manhattans plots of GWAS for meat quality traits. The SNPs on different chromosomes are denoted by different colors. The solid and dashed lines indicate the Bonferroni-corrected thresholds of the genome-wide and suggestive significances, respectively. The most significant SNP ss131569519 at 132.80 Mb on SSC15 for LM_pH24 h was close to the PRKAG3 gene with known effect on this trait
pH and drip loss
For pH and drip loss, a total of six suggestive SNPs were identified, including one on SSC1 for LM_pH45 min, three on SSC15 for LM_pH24 h, one on SSC17 for SM_ pH45 min and one on SSC4 for LM_DripEZ_24 h. No significant SNPs were found for pH45 min and DripEZ_24 h of SM.
Meat color and moisture content
We identified 60 and 42 SNPs associated with meat color and moisture content, respectively. Among them, 75 SNPs reached the genome-wide significance level, including 14 for LM_ColorM_a24 h, 14 for LM_ColorM_b24 h, 12 for SM_ColorM_a24 h, and 35 for LM_MoistureContent. On SSC12, the most significant SNP simultaneously associated with LM_ColorM_L24 h (P = 6.62e−06), LM_ColorM_a24 h (P = 1.97e−11) and LM_MoistureContent (P = 6.20e−12) was the SNP ss131468701 located at 57.95 Mb, which resides only about 100 Kb away from the other two genome-wide significant SNPs ss131468595 and ss131468514 for the LM_ColorM_b24 h and SM_ColorM_a24 h, respectively (Fig. 2). These significant SNPs accounted for substantial proportions of phenotypic variances, ranging from 10.8 to 24.8 % (Table 3). Besides SSC12, we also identified associated SNPs on other chromosomes for meat color and moisture content, such as one SNP on SSC18 for LM_ColorM_a24 h, one SNP on SSC4 and one on SSC1 for SM_ColorM_a24 h, one on SSC1 for SM_ColorScore_24 h and eight on SSC4 for LM_MoistureContent. The SNPs on SSC4 for LM_MoistureContent also reached the genome-wide significance level (P = 3.94e−07).

Magnitudes of correlations between the fifteen meat quality traits. The dots indicate the significant (P < 0.05) correlation coefficients between each pair of traits. Their sizes and colors, respectively, represent the degree and direction (positive and negative) of the correlation coefficient (Color figure online)

Haplotype structure of a 1-Mb region on SSC12 containing genome-wide significant SNPs associated with the LM_MoistureContent, LM_ColorM_L24 h and LM_ColorM_a24 h. Solid lines mark the identified block
Haplotype analyses
We focused our haplotype analysis on the most significant QTL region on SSC12 that spans 500 Kb upstream and downstream of the peak SNP ss131468701. The results were illustrated in Fig. 3 and Table 4. We identified two haplotype blocks in which SNPs were in high LD (r 2 > 0.8). Block 1 spanned 143 kb and contained five SNPs including the peak SNP ss131468701. Block 2 of 13 kb consisted of two SNPs. In the block 1, four distinct haplotypes (ACGAG, AAAGA, GAGGA, and GAAGA, referred to as Hap1, Hap2, Hap3, and Hap4 hereinafter) were identified with frequencies of 22, 18, 15, and 46 %, respectively. Hap2, Hap3, and Hap4 were significantly associated with low moisture content, high L* and a* values in LM (P < 0.05), while Hap1 displayed opposite effects on these traits (Table 4).
Discussion
Comparison of QTLs identified in this study with those in previous studies
The current GWAS results demonstrated 108 SNPs (24 unmapped) associated with the 10 meat quality traits. Recently, some authors including us have reported GWAS results for meat quality traits in different pig populations. We previously identified 11 significant SNPs and 116 suggestive SNPs associated with 25-related meat quality traits in a White Duroc × Erhualian F2 intercross and Chinese Sutai pigs (Ma et al. 2013). Most QTLs were located on SSC 2, 3, 4, and X for pH, on SSC 1, 3, 4, 5, and 15 for drip loss, on SSC 2, 4, 5, 6, 7, 10, 12, 13, and 14 for meat color, on SSC 7 and 10 for moisture content. Luo et al. (Luo et al. 2012) also detected a total of 45 significant SNPs associated with one or several meat quality traits in a Large White × Minzhu F2 intercross; of these SNPs, 36 were located in a region of approximately 12 Mb (between 43 and 55 Mb) on SSC12. In this study, we confirmed several QTLs (e.g., the SSC12 QTL) that were previously described in those crosses between Chinese and western domestic breeds, which suggests that the alleles at these loci have not fixed in breeds of Chinese or even Western origins. In addition, this study reveals eight novel loci: including ss131149663 on SSC1 for LM_pH45 min, ss107838737 on SSC17 for pH24 h, ss131468701 on SSC12 for LM_ColorM_L24 h, ss131065155 on SSC18 for LM_ColorM_a24 h, ss131468595 on SSC12 for LM_ColorM_b24 h, ss131241209 on SSC4, and ss131129540 on SSC1 for SM_ColorM_a24 h, ss131179086 on SSC1 for SM_ColorScore_24 h.
Comparison of trait-associated SNPs between LM and SM
Most scientific studies of meat focus on the LM for fresh meat consumption, while SM is also of interest because of its significance for cooked ham production. Table 2 illustrates the marked differences in the tested phenotype between LM and SM in the Laiwu pigs. As for genetic loci, except for the SSC12 QTL affecting meat color, no common QTL for a trait were shared by LM and SM, which was consistent with other studies. Biological mechanisms underlying the discrepancy have not been elucidated yet. Interestingly, remarkable differences have been observed when comparing the mRNA expression profiles of different pig muscles (Gallardo et al. 2012; Herault et al. 2014), and the differentially expressed genes mainly related to myogenesis process suggests dissimilar post-natal myogenic activity between the two muscles. Furthermore, from the applied perspective, differences in loci controlling meat quality between the two muscles are a relevant aspect to be taken into account when implementing genomic selection schemes devoted to improving these traits (Herault et al. 2014).
Possible pleiotropic QTLs
The present GWAS results showed two regions on SSC4 and SSC12 that were associated with more than one trait. The region between 56.22 and 61.49 Mb on SSC12 harbored a number of SNPs affecting five traits, including LM_ColorM_a24 h, LM_ColorM_b24 h, LM_ColorM_L24 h, LM_MoistureContent, and SM_ColorM_a24 h, which supports the findings of Luo et al. (2012) and Lee et al. (2012). The haplotypes in the QTL region also showed significant effects on the multiple traits. Since the correlation coefficients among these traits were significant (P < 0.05; Fig. 2), it is very likely that they were influenced by a common causative variant in the SSC12 region.
In addition, a region of 6.86 Mb (between 19.36 and 26.32 Mb) on SSC4 was found to be associated with both LM_DripEZ_24 h and LM_MoistureContent. Thus they may be controlled by the same causative gene on the SSC4 region.
Plausible candidate genes at the identified loci
Based on the GWAS results and information about gene function and expression pattern in muscle tissue, we recognized some interesting candidate genes nearby the association signals. It is known that skeletal muscle is a heterogeneous tissue composed of myofibers, adipose, connective, and vascular and nervous tissues (Herault et al. 2014). The structure and function diversity of contraction protein and metabolic protein cause the different skeletal muscle fiber type. Therefore, within the SSC12 QTL region around 57.95 Mb, the MYH1, MYH2, MYH3, and MYH13 genes, which belong to the myosin heavy chain gene family (MYH) and express highly in skeletal muscle, can be regarded as prime candidates. A targeted null mutation in MYH1 gene exhibit reduced growth, muscular weakness, kyphosis, and abnormal kinetics of muscle contraction and relaxation in mice (Leinwand et al. 1983; Skarnes et al. 2011). The Glu706Lys mutation in the MYH2 gene has been reported to be associated with a family congenital myopathy in human (Oldfors et al. 2004). For MYH3 gene, a structure–function analysis shows that nearly all of the MYH3 mutations are predicted to interfere with myosin’s catalytic activity and the mutations have been associated with two congenital contracture (arthrogryposis) syndromes, Freeman-Sheldon syndrome and Sheldon-Hall syndrome (Toydemir et al. 2006). So the four members of MYH gene family are all related to muscle development (Fan et al. 2011). The MAL2 gene is located close to the most significant SNPs ss131255131 for LM_MoistureContent and ss131256749 for LM_DripEZ_24 h on SSC4. This gene encodes a multispan transmembrane protein, which belongs to the MAL proteolipid family. This protein is a component of lipid rafts in polarized cells, and it primarily localizes to endosomal structures beneath the apical membrane (de Marco et al. 2002).
Three QTLs have been found for pH traits. The LPAR1 gene on SSC1 is likely to be responsible for LM_pH45 min. The integral membrane protein encoded by this gene is a lysophosphatidic acid (LPA) receptor from a group known as EDG receptors. These receptors are members of the G protein-coupled receptor superfamily. Utilized by LPA for cell signaling, EDG receptors mediate diverse biologic functions, including proliferation, platelet aggregation, and smooth muscle contraction (Moolenaar et al. 1997; Aziziyeh et al. 2009). The most significant SNP ss131569519 for LM_pH24 h(P = 1.72e−06)was detected at 132.80 Mb on SSC15. This QTL overlapped with a known QTL for meat pH values, glycogen content, glycolytic potential, reflectance, tenderness, and flavor score in Western commercial pig breeds, which has been ascribed to different structural or regulatory mutations in PRKAG3 gene (at 133.80 Mb) (Ciobanu et al. 2001; Otto et al. 2007; Ryan et al. 2012). No obvious candidate genes were found for LM_pH24 h on SSC17.
Conclusions
This GWAS revealed 75 genome-wide significant SNPs and 33 suggestively significant SNPs for 10 meat quality traits in Chinese purebred Laiwu pigs. We not only confirmed several previously reported QTLs but also presented some novel QTLs. Two QTL regions on SSC12 and SSC4 were found to affect more than one trait. A haplotype block of 143 Kb on SSC12 with five SNPs was identified and displayed four distinct haplotypes with significant association with moisture content and meat color. Several promising candidate genes close to significant SNPs were suggested to underlie the QTL effects. Our findings provide the first insights into the genetic basis of meat quality traits in Chinese indigenous pigs and pave the sound road to identification of causative variants, especially for the major loci on SSC12 and SSC4.
References
Andersson L, Haley CS, Ellegren H, Knott SA, Johansson M, Andersson K, Andersson-Eklund L, Edfors-Lilja I, Fredholm M, Hansson I et al (1994) Genetic mapping of quantitative trait loci for growth and fatness in pigs. Science 263:1771–1774
Aulchenko YS, Ripke S, Isaacs A, van Duijn CM (2007) GenABEL: an R library for genome-wide association analysis. Bioinformatics 23:1294–1296
Aziziyeh AI, Li TT, Pape C, Pampillo M, Chidiac P, Possmayer F, Babwah AV, Bhattacharya M (2009) Dual regulation of lysophosphatidic acid (LPA1) receptor signalling by Ral and GRK. Cell Signal 21:1207–1217
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263–265
Becker D, Wimmers K, Luther H, Hofer A, Leeb T (2013) A genome-wide association study to detect QTL for commercially important traits in Swiss large white boars. PLoS One 8:e55951
Berg EP (2006) Pork composition and quality assessment procedures, 1st edn. National Pork Producer Council (NPPC) Des Moines, IA, USA
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Breslow NE, Clayton DG (1993) Approximate inference in generalized linear mixed models. J Am Stat Assoc 88(421):9–25
Ciobanu D, Bastiaansen J, Malek M, Helm J, Woollard J, Plastow G, Rothschild M (2001) Evidence for new alleles in the protein kinase adenosine monophosphate-activated gamma(3)-subunit gene associated with low glycogen content in pig skeletal muscle and improved meat quality. Genetics 159:1151–1162
de Marco MC, Martin-Belmonte F, Kremer L, Albar JP, Correas I, Vaerman JP, Marazuela M, Byrne JA, Alonso MA (2002) MAL2, a novel raft protein of the MAL family, is an essential component of the machinery for transcytosis in hepatoma HepG2 cells. J Cell Biol 159:37–44
Druet T, Farnir FP (2011) Modeling of identity-by-descent processes along a chromosome between haplotypes and their genotyped ancestors. Genetics 188:409–419
Druet T, Georges M (2010) A hidden markov model combining linkage and linkage disequilibrium information for haplotype reconstruction and quantitative trait locus fine mapping. Genetics 184:789–798
Duan YY, Ma JW, Yuan F, Huang LB, Yang KX, Xie JP, Wu GZ, Huang LS (2009) Genome-wide identification of quantitative trait loci for pork temperature, pH decline, and glycolytic potential in a large-scale White Duroc x Chinese Erhualian resource population. J Anim Sci 87:9–16
Fan B, Onteru SK, Du ZQ, Garrick DJ, Stalder KJ, Rothschild MF (2011) Genome-wide association study identifies Loci for body composition and structural soundness traits in pigs. PLoS One 6:e14726
Fujii J, Otsu K, Zorzato F, de Leon S, Khanna VK, Weiler JE, O’Brien PJ, MacLennan DH (1991) Identification of a mutation in porcine ryanodine receptor associated with malignant hyperthermia. Science 253:448–451
Gallardo D, Pena RN, Quintanilla R, Ramirez O, Almuzara D, Noguera JL, Amills M (2012) Quantitative trait loci analysis of a Duroc commercial population highlights differences in the genetic determination of meat quality traits at two different muscles. Anim Genet 43:800–804
Herault F, Vincent A, Dameron O, Le Roy P, Cherel P, Damon M (2014) The longissimus and semimembranosus muscles display marked differences in their gene expression profiles in pig. PLoS One 9:e96491
Kim KS, Larsen N, Short T, Plastow G, Rothschild MF (2000) A missense variant of the porcine melanocortin-4 receptor (MC4R) gene is associated with fatness, growth, and feed intake traits. Mamm Genome 11:131–135
Lee KT, Lee YM, Alam M, Choi BH, Park MR, Kim KS, Kim TH, Kim JJ (2012) A whole genome association study on meat quality traits using high density SNP Chips in a cross between Korean native pig and landrace. Asian Aust J Anim Sci 25:1529–1539
Leinwand LA, Fournier RE, Nadal-Ginard B, Shows TB (1983) Multigene family for sarcomeric myosin heavy chain in mouse and human DNA: localization on a single chromosome. Science 221:766–769
Luo W, Cheng D, Chen S, Wang L, Li Y, Ma X, Song X, Liu X, Li W, Liang J, Yan H, Zhao K, Wang C, Wang L, Zhang L (2012) Genome-wide association analysis of meat quality traits in a porcine large white x Minzhu intercross population. Int J Biol Sci 8:580–595
Ma J, Ren J, Guo Y, Duan Y, Ding N, Zhou L, Li L, Yan X, Yang K, Huang L, Song Y, Xie J, Milan D, Huang L (2009) Genome-wide identification of quantitative trait loci for carcass composition and meat quality in a large-scale white duroc x Chinese Erhualian resource population. Anim Genet 40:637–647
Ma J, Yang J, Zhou L, Zhang Z, Ma H, Xie X, Zhang F, Xiong X, Cui L, Yang H, Liu X, Duan Y, Xiao S, Ai H, Ren J, Huang L (2013) Genome-wide association study of meat quality traits in a white duroc x Erhualian F2 intercross and Chinese Sutai pigs. PLoS One 8:e64047
Ma J, Yang J, Zhou L, Ren J, Liu X, Zhang H, Yang B, Zhang Z, Ma H, Xie X, Xing Y, Guo Y, Huang L (2014) A splice mutation in the PHKG1 gene causes high glycogen content and low meat quality in pig skeletal muscle. PLoS Genet 10:e1004710
Milan D, Jeon JT, Looft C, Amarger V, Robic A, Thelander M, Rogel-Gaillard C, Paul S, Iannuccelli N, Rask L, Ronne H, Lundstrom K, Reinsch N, Gellin J, Kalm E, Roy PL, Chardon P, Andersson L (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288:1248–1251
Moolenaar WH, Kranenburg O, Postma FR, Zondag GC (1997) Lysophosphatidic acid: G-protein signalling and cellular responses. Curr Opin Cell Biol 9:168–173
Nonneman DJ, Shackelford SD, King DA, Wheeler TL, Wiedmann RT, Snelling WM, Rohrer GA (2013) Genome-wide association of meat quality traits and tenderness in swine. J Anim Sci 91:4043–4050
Oldfors A, Tajsharghi H, Darin N, Lindberg C (2004) Myopathies associated with myosin heavy chain mutations. Acta Myol 23:90–96
Otto G, Roehe R, Looft H, Thoelking L, Knap PW, Rothschild MF, Plastow GS, Kalm E (2007) Associations of DNA markers with meat quality traits in pigs with emphasis on drip loss. Meat Sci 75:185–195
Pearson TA, Manolio TA (2008) How to interpret a genome-wide association study. JAMA 299:1335–1344
Ponsuksili S, Murani E, Trakooljul N, Schwerin M, Wimmers K (2014) Discovery of candidate genes for muscle traits based on GWAS supported by eQTL-analysis. Int J Biol Sci 10:327–337
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81:559–575
Ramos AM, Crooijmans RP, Affara NA, Amaral AJ, Archibald AL, Beever JE, Bendixen C, Churcher C, Clark R, Dehais P, Hansen MS, Hedegaard J, Hu ZL, Kerstens HH, Law AS, Megens HJ, Milan D, Nonneman DJ, Rohrer GA, Rothschild MF, Smith TP, Schnabel RD, Van Tassell CP, Taylor JF, Wiedmann RT, Schook LB, Groenen MA (2009) Design of a high density SNP genotyping assay in the pig using SNPs identified and characterized by next generation sequencing technology. PLoS One 4:e6524
Rassmussen AJ, Andersson M (1996) New method for determination of drip loss in pork muscles. In: Proceedings 42nd international congress of meat science and technology. Lillehmmer, Norway, pp 286–287
Ren J, Duan Y, Qiao R, Yao F, Zhang Z, Yang B, Guo Y, Xiao S, Wei R, Ouyang Z, Ding N, Ai H, Huang L (2011) A missense mutation in PPARD causes a major QTL effect on ear size in pigs. PLoS Genet 7:e1002043
Riquet J, Gilbert H, Servin B, Sanchez MP, Iannuccelli N, Billon Y, Bidanel JP, Milan D (2011) A locally congenic backcross design in pig: a new regional fine QTL mapping approach miming congenic strains used in mouse. BMC Genet 12:6
Rosenvold K, Andersen HJ (2003) Factors of significance for pork quality-a review. Meat Sci 64:219–237
Rothschild M, Bidanel JP (2002) Current status of quantitative trait locus mapping in pigs. Pig News Inf 23:39N–54N
Ryan MT, Hamill RM, O’Halloran AM, Davey GC, McBryan J, Mullen AM, McGee C, Gispert M, Southwood OI, Sweeney T (2012) SNP variation in the promoter of the PRKAG3 gene and association with meat quality traits in pig. BMC Genet 13:66
Sanchez MP, Riquet J, Iannuccelli N, Gogue J, Billon Y, Demeure O, Caritez JC, Burgaud G, Feve K, Bonnet M, Pery C, Lagant H, Le Roy P, Bidanel JP, Milan D (2006) Effects of quantitative trait loci on chromosomes 1, 2, 4, and 7 on growth, carcass, and meat quality traits in backcross Meishan x large white pigs. J Anim Sci 84:526–537
Sanchez MP, Tribout T, Iannuccelli N, Bouffaud M, Servin B, Tenghe A, Dehais P, Muller N, Del Schneider MP, Mercat MJ, Rogel-Gaillard C, Milan D, Bidanel JP, Gilbert H (2014) A genome-wide association study of production traits in a commercial population of large white pigs: evidence of haplotypes affecting meat quality. Genet Sel Evol 46:12
Sartelet A, Druet T, Michaux C, Fasquelle C, Geron S, Tamma N, Zhang Z, Coppieters W, Georges M, Charlier C (2012) A splice site variant in the bovine RNF11 gene compromises growth and regulation of the inflammatory response. PLoS Genet 8:e1002581
Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474:337–342
Stratz P, Wellmann R, Preuss S, Wimmers K, Bennewitz J (2014) Genome-wide association analysis for growth, muscularity and meat quality in Pietrain pigs. Anim Genet 45:350–356
Suzuki K, Irie M, Kadowaki H, Shibata T, Kumagai M, Nishida A (2005) Genetic parameter estimates of meat quality traits in Duroc pigs selected for average daily gain, longissimus muscle area, backfat thickness, and intramuscular fat content. J Anim Sci 83:2058–2065
Toydemir RM, Rutherford A, Whitby FG, Jorde LB, Carey JC, Bamshad MJ (2006) Mutations in embryonic myosin heavy chain (MYH3) cause freeman–sheldon syndrome and Sheldon–Hall syndrome. Nat Genet 38:561–565
Uimari P, Sironen A (2014) A combination of two variants in PRKAG3 is needed for a positive effect on meat quality in pigs. BMC Genet 15:29
Van Laere AS, Nguyen M, Braunschweig M, Nezer C, Collette C, Moreau L, Archibald AL, Haley CS, Buys N, Tally M, Andersson G, Georges M, Andersson L (2003) A regulatory mutation in IGF2 causes a major QTL effect on muscle growth in the pig. Nature 425:832–836
Wellcome Trust Case Control C (2007) Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447:661–678
Yang Q, Cui J, Chazaro I, Cupples LA, Demissie S (2005) Power and type I error rate of false discovery rate approaches in genome-wide association studies. BMC Genet 6(Suppl 1):S134
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Zhang F, Zhang Z, Yan X, Chen H, Zhang W, Hong Y, Huang L (2014) Genome-wide association studies for hematological traits in Chinese Sutai pigs. BMC Genet 15:41
Acknowledgments
This research was supported by the Key Project of National Nature Science Foundation of China (No. 31230069), the Development Programs for Basic Research of China (973 Programs, Nos. 2012CB722502 and 2012CB124702) and National High-Tech Research and Development Program of China (2013AA102502).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Xinwei Xiong and Xianxian Liu have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xiong, X., Liu, X., Zhou, L. et al. Genome-wide association analysis reveals genetic loci and candidate genes for meat quality traits in Chinese Laiwu pigs. Mamm Genome 26, 181–190 (2015). https://doi.org/10.1007/s00335-015-9558-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-015-9558-y