
Abstract
Key message
The heat stress transcription factor HSFA2e regulates both temperature and drought response via hormonal and secondary metabolism alterations.
Abstract
High temperature and drought are the primary yield-limiting environmental constraints for staple food crops. Heat shock transcription factors (HSF) terminally regulate the plant abiotic stress responses to maintain growth and development under extreme environmental conditions. HSF genes of subclass A2 predominantly express under heat stress (HS) and activate the transcriptional cascade of defense-related genes. In this study, a highly heat-inducible HSF, HvHSFA2e was constitutively expressed in barley (Hordeum vulgare L.) to investigate its role in abiotic stress response and plant development. Transgenic barley plants displayed enhanced heat and drought tolerance in terms of increased chlorophyll content, improved membrane stability, reduced lipid peroxidation, and less accumulation of ROS in comparison to wild-type (WT) plants. Transcriptome analysis revealed that HvHSFA2e positively regulates the expression of abiotic stress-related genes encoding HSFs, HSPs, and enzymatic antioxidants, contributing to improved stress tolerance in transgenic plants. The major genes of ABA biosynthesis pathway, flavonoid, and terpene metabolism were also upregulated in transgenics. Our findings show that HvHSFA2e-mediated upregulation of heat-responsive genes, modulation in ABA and flavonoid biosynthesis pathways enhance drought and heat stress tolerance.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The food security for global population is solely dependent on crop production which is determined by the variations in climate. The high variations in climatic factors lead to a reduction in crop production and severely impact food supply (Ray et al. 2015). Temperature and water availability primarily determine the crop yield and the fluctuations beyond optimal range impose deleterious effects on yield potentials. It has been reported if the rise in global surface temperature continues at the current rate, then it will increase up to or beyond 1.5 °C in comparison to the pre-industrial climate by ~2040 and the warming will be higher on land than that of ocean surface (Masson-Delmotte et al. 2019). More than half of the calorific requirement of world population is fulfilled by cereal crops (Nelson et al. 2010), which are highly susceptible to heat stress during the reproductive phase (Farooq et al. 2011). The combined effect of heat and drought stress is comparatively more damaging to crop yield in comparison to the individual impact of either of the stresses (Lamaoui et al. 2018; Yashavanthakumar et al. 2021). Crop improvement is an essential step to ascertain food security under future warmer climate with water-limiting conditions (Atlin et al. 2017). It is more challenging for plant scientists to increase the crop yield as well as abiotic stress tolerance, simultaneously, to cope up with the threat of global climate change (Battisti and Naylor 2009).
Under abiotic stress, one of the major obstacles for plants in maintaining the yield is to overcome the excessive generation and increased accumulation of ROS. The compromised ROS homeostasis results in oxidizing environment in plant cells. To avoid the oxidative damages, plants have evolved the molecular strategies in the form of stress responses. As plants cannot move and migrate to avoid unfavorable environmental conditions, they face stress conditions and develop tolerance/resistance to survive. To cope with environmental challenges, plants have evolved an intricate response system; one of the primary actions that improve stress tolerance is dynamic reprogramming of transcriptional activity. Under various stress circumstances, several transcription factor families are implicated in complex and overlapping responses. The major transcriptional regulators which regulate and reprogram the gene expression under high-temperature and drought stress are HSFs, WRKY, MYB, DREB, MBF1C (multiprotein bridging factor 1c), bZIP (basic region/leucine zipper motif), ERF (ethylene-responsive factor), and NAC. Plant heat shock response (HSR) is regulated by heat stress transcription factors (HSFs). The DNA-binding ability of HSFs allow them to bind with a consensus cis-regulatory sequence nAGAAnTTCT (Heat shock element—HSE) present in the promoter of heat-responsive genes (Nover et al. 1996; Mishra et al. 2020). Predominant upregulation of class A HSFs in response to high temperature has been widely characterized. Among the class A, HSFA2 gene showed strong inducibility in both dicot as well as monocot plants against heat shock (Charng et al. 2007; Giorno et al. 2010; Xue et al. 2014; Chauhan et al. 2011). Schramm et al. (2006) reported that the expression of ascorbate peroxidase and galactinol synthase is also regulated by HSFA2 under heat stress. In HSR, HSFs directly regulate the transcription of HSPs, antioxidant enzymes, and other thermotolerance contributing genes (Lämke et al. 2016; Wang et al. 2017). Thus, the molecular pathways in plants are precisely reprogramed under the regulation of stress-responsive transcription factors to minimize the damage and maintain the physiological processes.
Transcriptome-based studies showed that the expression of plant HSF genes shows modulation not only under high temperature but also during other abiotic stresses. Moreover, the functional characterization of various HSF genes showed their role in transcriptional regulation of different genes which are responsive to drought, salinity, high light, cold and heavy metal stresses. Similarly, the transcriptome profiles under different stress conditions show overlapping in gene expression patterns suggesting the cross-talk among these stress responses (Swindell et al. 2007). HSF genes are induced by multiple stress conditions; most of the abiotic stresses ultimately result in oxidative stress, which arise due to excessive generation and accumulation of ROS in plant cell. Excessive ROS is harmful to physiological processes of plants, though ROS-mediated signaling plays an important role in evoking the plant defense system (Mittler et al. 2022). Previous studies showed that ROS generating conditions induce the expression of HSF genes (Pérez-Salamo et al. 2014; Choi et al. 2019). It has been observed that the overexpression of single HSF gene may have impact on tolerance against multiple abiotic stresses. Not only the thermotolerance but drought, water stress, salinity, and heavy metal stress tolerance were also enhanced in transgenic plants overexpressing the candidate genes from HSF Class A1 and A2 (Li et al. 2005, 2014; Bechtold et al. 2013).
To maintain optimum yield potential in the projected adverse climate, transgenic cereal plants can be developed with altered HSF expression levels. These transgenic plants will be able to tolerate adverse environmental conditions such as high temperature, water depletion, and saline soil, ultimately resulting in ensuring food security for the global population. Among the 23 barley HSFs, HvHSFA2e was not only the highest expressing HSF under 30 min of heat shock (HS) but also one of the early heat-responsive HSFs, showing heat-induced upregulation as early as 10 min of HS onset (Mishra et al. 2020). Considering these characteristics, HvHSFA2e gene was targeted for constitutive overexpression to reveal its potential role in enhancing the abiotic stress tolerance in barley plants in the present study.
Materials and methods
Plant material and growth conditions
Seeds of barley (Hordeum vulgare L.) Cv. Golden promise were germinated in a mixture of soil and soilrite (1:1) under controlled conditions in greenhouse 18/16 ℃ (day/night) temperature and 16 h photoperiod. 2-week-old wild-type (WT) plants were used to assess the stress inducible expression of HvHSFA2e gene. For heat shock (HS), tissue samples (Shoot and root) were harvested at 0 (control), 30 min, and 2 h time points from the plants exposed to 42 ℃ temperature. For drought stress, 2-week-old plants were exposed to drought stress by stopping the water supply for next 2 weeks. The shoot and root samples were harvested from 4-week-old plants grown under water-limiting conditions. For drought stress experiment, control tissue was collected from 4-week-old plants growing under optimal non-stress conditions. Harvested stress-exposed plant tissue samples were snap frozen in liquid nitrogen and used for RNA isolation.
For genetic transformation of barley, immature embryos harvested from field as well as greenhouse grown plants were used as explant.
Transgenic lines and WT plants were grown in above-described conditions, 2-week-old plants were subjected to 40 °C for 4 h to assess the thermotolerance. For drought tolerance analysis, 2-week-old transgenic and WT plants were exposed to drought stress by limiting the water supply for next 2 weeks.
Cloning of HvHSFA2e cDNA into binary vector and genetic transformation of barley
A 2022 bp DNA fragment corresponding to full-length cDNA (FL- cDNA) of HvHSFA2e gene (AK358380) was amplified from the plasmid provided by GeneBank, NARO, Japan using gene specific primer pair (Supplementary Table S1). The PCR product was cloned in PCR cloning vector pJET1.2 (Invitrogen, Thermofisher Scientific USA) and further subcloned in binary vector p6oAct-UbiZm-LH (DNA cloning services, Hamburg Germany). A restriction digestion was performed with SwaI and EcoRV enzymes to obtain the ZmUbi1:HvHsfA2e:NosT cassette. This cassette was cloned in modified gateway binary vector pANIC6B (Mann et al. 2012) and used to transform Agrobacterium strain EHA105. Agrobacterium-mediated genetic transformation of barley was performed according to Hensel et al. (2009) to develop the HvHSFA2e overexpression lines (O.E.).
Transactivation assay
The yeast system was used for assessing the transactivation potential of HvHsfA2e protein. The HvHSFA2e ORF was amplified using the ORF specific primer pair (Supplementary Table S1) and fused to GAL4 BD in the yeast vector pGBKT7 (Clontech, CA, USA), generating the constructs pGBKT7:HvHsfA2e. The empty pGBKT7 was used as a negative control. Lithium acetate-polyethylene glycol-mediated transformation procedure was followed for transferring the pGBKT7:HvHsfA2e plasmid into yeast strain AH109. The transformants were allowed to grow in SD medium lacking amino acid tryptophan and histidine at 30°C. To assess if there was leaky expression of HIS3 reporter gene, dilutions were dotted on the respective dropout media supplemented with 1 mM and 5 mM of 3 AT. Each experiment was performed three times.
Subcellular localization through transient expression in Nicotiana benthamiana
The ORF of HvHSFA2e gene without stop codon was amplified using the HvHSFA2e-GFP-F and HvHSFA2e-GFP-R primer pair with flanking attB sites (Supplementary Table S1). The PCR product was cloned into pDONR221 by Gateway™ BP Clonase™ II Enzyme mix (Invitrogen), and a LR reaction was performed with pB7FWG2 empty vector and pDONR221-HvHSFA2e with LR Clonase™ enzyme mix (Invitrogen) to generate the construct pB7FWG2:35S:HvHSFA2e:GFP. The vector pB7FWG2-35S:HvHSFA2e:GFP was transformed into A. tumefaciens GV3101. Transient expression in N. benthamiana through Agrobacterium infiltration was performed according to Li, (2011). Protein localization was analyzed 24–48 h after infiltration by confocal laser scanning microscope (TCS SP8, Leica). GFP was excited at 488 nm and the fluorescence detected between 498 and 525 nm.
Confirmation and transgene expression analysis of HvHSFA2e overexpression line
For the confirmation of successful integration of targeted gene, leaf tissue of putative transgenic plants was used to isolate the genomic DNA using DNA-XPress™ Reagent (MB501, HiMedia Laboratories, Mumbai, India) as per manufacturer’s instructions. Gene-specific primers amplifying the selection marker gene Hygromycin phosphotransferase (HPT Full F-R), and reporter gene β-glucuronidase (Gusplus) were used to confirm the integration event in barley transgenic lines (Supplementary Table S1).
The expression level of HvHSFA2e gene in transgenic lines was analyzed through qRT-PCR. The expression analysis was done using QuantStudio 3 system and PowerUp SYBR Green Master Mix (Applied Biosystems, Thermo Fisher Scientific, USA) according to the manufacturer’s instructions and primer Express 3.0 software (Thermo Scientific, USA) was used to design the primers. Leaf tissue of WT barley plant grown under control conditions was used as control and snoR14 gene was used as internal reference control (Ferdous et al. 2015). The fold change calculation for gene expression was done using the formula 2−ΔΔCt (Livak and Schmittgen 2001). Similarly, the increased expression of other genes, which were found upregulated in RNA-seq data, was validated using the qRT-PCR.
Detection of GUS activity
Histochemical Gus assay was performed to confirm the stable transformation. The transformed immature embryos and leaf tissue of mature transgenic plants were subjected to Gus assay. The transgenic material was transferred in a microcentrifuge tube and immersed in freshly prepared X-gluc solution containing 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide, and 0.1% Triton X-100, followed by vacuum infiltration for 10 min and overnight incubation at 37℃. All samples were washed and fixed in 70% ethanol to remove the photosynthetic pigments prior to visualize the staining and taking photographs. GUS expression was observed under a stereo zoom microscope.
Histochemical detection, visualization, and quantification of ROS
The formation of superoxide free radicals (O2•−) in leaves was detected by NBT staining method (Wohlgemuth et al. 2002). The leaves sections were immersed in NBT-PBS solution and incubated at room temperature under dark conditions until the blue spot of formazan appeared.
The formation and accumulation of peroxide radical in leaves was detected by DAB staining method (Thordal-Christensen et al. 1997). The leaf sections were submerged in 1 mg/mL DAB–HCl (pH3.8) for 12 h under light condition. The peroxide accumulation sites can be visualized as the brown spots generated due to the polymerization of DAB. All samples were washed and fixed in 70 % ethanol to remove the pigments prior to visualize and taking photographs.
The leaf H2O2 content was quantified according to Chakraborty et al. (2016) using Amplex™ Red Hydrogen Peroxide/Peroxidase Assay Kit (Thermo Fisher Scientific, USA).
Determination of enzymatic antioxidant activities
To determine the activities of antioxidative enzymes, 500 mg leaf tissue was homogenized in 5 mL of assay buffer containing 100 mM phosphate buffer pH7.0, 2mM Na2EDTA, 1% PVP. The solution was centrifuged at 15,000×g for 20 min at 4 °C. The clear supernatant was collected and kept at 4 °C to use as crude enzyme extract in estimation protocols.
The ascorbate peroxidase activity was assayed according to Nakano and Asada (1981). The assay mixture consisted of 150 μL of the enzyme extract, 50 mM phosphate buffer (pH 7.0), 0.2 μM EDTA, 0.2 mM ascorbate, and 2.0 mM H2O2 in a total volume of 3 mL. Ascorbate oxidation was monitored by reading the absorbance at 290 nm at the moment of H2O2 addition and 1 min later. The difference in absorbance (ΔA290) was divided by the ascorbate molar extinction coefficient (2.8 mM–1.cm–1) and the enzyme activity expressed as μmol of H2O2 min–1 mg–1 protein, considering that 1.0 mol of ascorbate reduces 1.0 mol of H2O2 (McKersie and Leshem 1994).
The activity of superoxide dismutase was determined by measuring its ability to inhibit the photochemical reduction of nitro blue tetrazolium chloride, as described by Giannopolitis and Ries (1977). The assay mixture consisted of 300 μL of the enzyme extract, 100 mM phosphate buffer (pH 7.8), 3 mM EDTA, 200 μM methionine, 2.25 μM nitro blue tetrazolium, and 60 μM riboflavin (added at last) in a total volume of 3.2 mL. The tubes were inverted for 2–3 times for proper mixing and placed under fluorescent light. After 15 min, lights were switched off to stop the reaction. Then the tubes were immediately put in dark. Absorbance was measured at 560 nm. SOD activity was estimated by the method given by Fridovich (1986), and unit activity was expressed as enzyme utilized for 50% inhibition of NBT reduction.
Estimation of chlorophyll and carotenoid content, relative water content, solute leakage, and lipid peroxidation
For estimation of chlorophyll and carotenoid pigments, 500 mg fresh leaf tissue was homogenized in 5 mL of 80 % acetone and centrifuged at 6000 rpm for 15 min. The supernatant was collected and absorbance was measured at 480, 510 nm for carotenoid content and 645, 663 nm for chlorophyll a & b. The estimation of photosynthetic pigments was done according to Lichtenthaler and Wellburn (1983).
The relative water content (RWC) displays the hydration status of leaves. Freshly harvested leaf sections of known weight were kept in sterile distilled water for 24 h to obtain the turgid weight. The samples were then kept at 65 °C in a hot air oven until a constant weight was achieved to calculate the dry weight. RWC was calculated as RWC (%) = [(FW − DW)/(TW − DW)] × 100 (Ings et al. 2013), where FW indicates fresh weight, DW indicates dry weight, and TW indicates turgid weight.
The extent of solute leakage is used to determine the membrane stability in plants under unfavorable conditions. To compare the membrane stability of wild-type and transgenic lines, the solute leakage percentage was determined according to Dionisio-Sese and Tobita (1998) under control and heat stress conditions.
The lipid peroxidation in plants under stressed condition is measured in the form of MDA content which was estimated according to the method described by Zheng et al. (2012). The thiobarbituric acid (TBA) reaction was used to quantify the malondialdehyde (MDA) level, a byproduct of lipid peroxidation. 5 mL of 5% trichloroacetic acid (TCA) was added to 0.5 g of fresh leaf sample, and the mixture was homogenized. The homogenized mixture was centrifuged for 20 min at 10,000 rpm. Four milliliters of 0.5% thiobarbituric acid (TBA) were added to one milliliter of supernatant, and the mixture was heated for 30 min on water bath until an orange–yellow hue developed. Thereafter, it was quickly cooled on ice and absorbance was measured at 532 nm. MDA has an extinction coefficient of 155.
RNA-seq and data analysis
RNA isolation from leaf tissues of 5-week-old non-transformed wild-type and HvHSFA2e overexpression transgenic lines was done using RNeasy plant RNA isolation kit as per manufacturer’s instruction (Qiagen, Germany; Cat No./ID: 74904 and 79254). High-quality RNA samples were outsourced to Bionivid Technology Private Limited (Bengaluru, India) for RNA-seq using Illumina HiSeq platform. The RNA-seq data (accession number GSE241562) used and analyzed during this study are available in the GEO, NCBI database.
Quality control of all the reads obtained for each sample was done using NGSQC Tool kit (Patel et al. 2012) and reads with Phred score >Q20 were selected for further analysis. Barley reference genome was downloaded from Ensembl Plants database (http://ftp.ensemblgenomes.org/pub/plants/release-53/fasta/hordeum_vulgare/) for alignment and mapping of reads using STAR pipeline (Dobin et al. 2013; https://github.com/alexdobin/STAR) and Trinity platform (genome guided mode) (Haas et al. 2013; https://github.com/trinityrnaseq/trinityrnaseq). Abundance estimation was calculated using RSEM (RNA-Seq by Expectation-Maximization) method (Li and Dewey 2011), and differential gene expression analysis was performed using DESeq2 (Love at al. 2014; https://bioconductor.org/packages/release/bioc/html/DESeq2.html). Transcripts having log2 fold change greater ≥ 2 were considered as DEGs. Volcano plots were constructed using Plotly (pandas) in python (https://plotly.com/python/volcano-plot/#volcanoplot). Functional annotation of the transcriptome was performed using the Trinotate pipeline (https://github.com/Trinotate/Trinotate.github.io/blob/master/index.asciidoc), employing uniport-swissprot (https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/) database for blastx and blastp searches. Cytoscape (v 3.9.1) plug-in bingo (Maere et al. 2005) was used to perform GO (Gene Ontology) enrichment analysis. Enriched GO terms were identified separately for upregulated and downregulated sets of genes in barley transgenics as compared to wild type. Obtained enriched terms were plotted using the web server Revigo (http://revigo.irb.hr/; Supek et al. 2011). Heatmaps were constructed using ggplots (Warnes et al. 2020) and RColorBrewer (Neuwirth 2014) packages in R.
Statistical analysis
In present study, all the data were generated by three independent experiments; and each experiment was performed with three technical replicates. Microsoft Office Excel 2021 was used to perform the statistical analysis. Error bars in the graphs denote standard errors of the mean values (±SE). The statistical significance between WT and transgenic data sets was determined by student’s t test. The level of significant difference was set at P < 0.05 (P-value; * = < 0.05; **= < 0.01; ***= < 0.001).
Results
Generation of transgenic barley lines overexpressing HvHSFA2e and subcellular localization of HvHSFA2e protein
The HSF gene family of barley possess 23 members, among these genes, HvHSFA2e showed highest expression under heat stress conditions in vegetative shoot of barley cultivar RD2786 (Mishra et al. 2020). HvHSFA2e gene encodes for 371 amino acid long protein with 41.1 kDa molecular weight. It displays all characteristic features of a class A HSF, including conserved HSF domain or DNA-binding domain (DBD), oligomerization domain, transactivation domain (AHA domain), and nuclear localization signal (NLS) (Fig. 1A). We analyzed the transactivation potential of HvHSFA2e through transactivation assay. The yeast strain AH109 cells transformed with the pGBKT7:HvHSFA2e, grew well on the medium of SD/−Trp, and the colonies appeared blue on X-α-Gal supplemented SD/−Trp medium, revealing that HvHsfA2e possess transactivation potential (Fig. 1B). Similar to Indian commercial barley cultivar RD2786, the HvHSFA2e gene of European spring barley cultivar Golden promise (suitable for tissue culture) showed the heat-shock-induced expression in shoot tissue (Fig. 1C).

HvHSFA2e gene is highly heat-inducible and encoded protein shows transactivation potential and nuclear–cytoplasmic localization. HvHSFA2e is a class A HSF, consisting 371 amino acids. It displays the presence of HSF domain (DNA-binding domain), oligomerization domain, transactivation domain, and nuclear localization signal (A), transactivation potential of HvHsfA2e in GAL4BD filter lift assay (B), qRT-PCR-based assessment showed the heat and drought inducible expression of HvHSFA2e gene in WT seedlings; heat shock (HS) exposure was given at 42 °C for 30 min., 2 h and drought exposure was given for 2 weeks. snoR14 gene was used as internal control reference for calculating the expression level in terms of relative fold changes by the ΔΔCt method (C), HvHSFA2e ORF was cloned under CaMV35S promoter, C-terminal GFP fused construct GFP:HvHSFA2e transiently expressing in N. benthamiana leaves was found to be localized in nucleus and cytoplasm (D). (HS heat shock, DBD DNA binding domain, HR-A/B hydrophobic repeats A and B, NLS nuclear localization signal, AHA aromatic–hydrophobic-acidic amino acid domain)
The Agrobacterium-mediated genetic transformation was performed to generate the HvHSFA2e O.E. in barley Cv. Golden promise. The HvHSFA2e was cloned under the regulation of Ubiquitin promoter of Zea mays (ZmUbi promoter) in modified pANIC6B binary vector for its constitutive expression. Agrobacterium-mediated transformation of barley immature embryos through vacuum infiltration (Hensel et al. 2009) followed by the tissue culture resulted into the generation of the overexpression transgenic lines.
The putative 17 T0 transgenic lines were confirmed by the PCR amplification of Gusplus (reporter) and Hpt (selectable marker) genes. The positive results of PCR reactions using the genomic DNA isolated from T0 plants confirmed the successful-stable transformation. The primers used for PCR reactions were specific to full-length Gusplus (FL Gusplus) and Hpt genes present in pANIC6B binary vector. In PCR analysis, all the 17 T0 plants showed positive results for amplification of targeted genes; HptII and Gusplus. The bands of desired size were visualized on the agarose gel (Supplementary figure 2 A and B).
The expression of Gusplus reporter gene was assessed through histochemical analysis at different stages of tissue culture as well as in mature transgenic lines. The callus, regenerants as well as the mature plants of T0 progeny showed the expression of reporter gene (Supplementary figure 3). Further, expression of HvHSFA2e was assessed till T2 progeny and the TR4 and TR5 lines which showed stable constitutive expression were selected for biochemical and transcriptome analysis (Supplementary Figure 4).
The subcellular localization of HvHSFA2e protein was analyzed by constructing a fusion gene with GFP at C-terminal and transiently expressing it in N. benthamiana leaves. The confocal laser scanning microscopy showed the uniform localization of fusion protein in cytoplasm as well as nucleus, suggesting the cytoplasmic and nuclear localization of the HvHSFA2e protein (Fig. 1D).
Overexpression of HvHSFA2e resulted in reduced growth rate and increased photosynthetic pigment formation in seedlings
The HSF genes have been reported for their role in plant growth and development. To investigate the effect of HvHSFA2e overexpression on plant growth, we observed the growth pattern of WT and transgenic lines (15 plants each) under controlled optimal conditions. The transgenic lines showed slow shoot and root growth during seedling and early vegetative phase in comparison to WT plants. The 10-day-old transgenic seedlings showed significant reduction in growth rate in the form of reduced shoot and root length (TR4: shoot-9.0 cm, root-4.8 cm; TR5: shoot-10.3 cm, root-5.3 cm) when compared to WT seedlings (Shoot-14.1 cm, root–7.04 cm) (Fig. 2A, B, and C). However, there were no phenotypic differences in WT and transgenic plants grown under control conditions during late vegetative and reproductive phase (data not shown).

Plant growth and photosynthetic pigments. After 10 days of germination, the impact of HvHSFA2e overexpression on seedling growth was observed. WT and HvHSFA2e overexpression lines were grown under optimal conditions (18/16 °C day/night temperature and 16 h photoperiod) for 10 days and photographed for morphological assessment (A), the graphs show the average shoot and root length of 15 seedling each WT and transgenic (TR4, TR5) lines (B,C), transgenic plants show increased chlorophyll and carotenoid content in comparison to WT (D). Data represent mean ± SE. Asterisks (*, **, ***) were used to indicate significant differences between WT and transgenic lines calculated using Student’s t test, *P value < 0.05, **P value < 0.01, and ***P value < 0.001
The estimation of photosynthetic pigments in WT and transgenic plant leaves showed that the total chlorophyll and carotenoid contents in transgenic plants were significantly higher (TR4—Chl: 1.75, carotenoid: 0.704; TR5—Chl: 1.77, carotenoid: 0.71 mg/g FW) than that in WT (total Chl: 1.18, carotenoid: 0.399 mg/g FW) plants (Fig. 2D), indicating an enhanced synthesis of photosynthetic pigments in HvHSFA2e O.E.
Overexpression of HvHSFA2e improves the activity of antioxidant enzymes and reduces the ROS accumulation and lipid peroxidation
To protect the plants from oxidative damage, the antioxidant enzymes determine the concentration of ROS species in plant cells by eliminating the excessive accumulation of ROS. Superoxide dismutase (SOD) and ascorbate peroxidase (APX) are the two major components of enzymatic antioxidant defense system. The activities of these two enzymes were measured in transgenic lines (TR4 and TR5) as well as wild-type plants under control conditions. The transgenic lines showed significantly enhanced activity for both the enzymes in comparison to wild-type barley plants (Fig. 3A, B).

Antioxidant enzyme activity and ROS accumulation analysis. Activity of ROS-scavenging enzymes viz APX and SOD in WT and transgenic lines (TR4 and TR5) (A, B), visualization of Superoxide radical accumulation by NBT staining (C) and peroxide radical accumulation by DAB staining (D) in the leaves of wild-type (WT) and HvHSFA2e overexpression lines (TR4 and TR5) under control and HS conditions. Data represent mean ± SE. Asterisks (*, **, ***) represent significant differences between WT and transgenic lines calculated using Student’s t test, *P value < 0.05, **P value < 0.01, and ***P value < 0.001
The high temperature stress causes increased generation and accumulation of ROS species. The unchecked accumulation of ROS species results in oxidative damage to plant cells at molecular and physiological level. The accumulation of peroxide and superoxide anions was investigated in the leaf tissue of wild type as well as transgenic lines (TR4 and TR5) under control and heat stress conditions. The accumulation of superoxide free radical in leaf tissue was displayed through the NBT staining (Wohlgemuth et al. 2002), whereas the peroxide radical accumulation was visualized by DAB staining (Liu and Friesen 2012). Under control conditions, no significant difference was observed in ROS accumulation in transgenic lines and wild-type plants; however, the transgenic lines displayed significantly less accumulation of peroxide as well as superoxide anions in leaf tissue in comparison to wild-type barley plants under heat stress (Fig. 3C, D).
Overexpression of HvHSFA2e improves the thermotolerance in transgenic barley plants
To investigate the impact of HvHSFA2e overexpression on plant thermotolerance, we analyzed the performance of transgenic lines under heat stress. Two-week-old WT and transgenic plants were subjected to a heat shock at 40 °C for 5 h, WT plants displayed severe leaf damage in the form of wilting, whereas transgenic lines showed resistance in comparison to the WT (Fig. 4A, B). Moreover, the quantification of H2O2 content in leaf tissue also revealed the more deleterious impact on WT plants in comparison to HvHSFA2e O.E., as under heat stress conditions, H2O2 content was significantly higher in WT plants (14.41 µmol/g FW) in comparison to transgenic lines (TR4-7.86, and TR5-8.04 µmol/g FW) (Fig. 4 C).

Physiological response of wild-type (WT) and HvHSFA2e overexpression lines under heat stress. 2-week-old WT and overexpression lines were subjected to heat stress (40 °C for 4 h), WT plants show comparatively more negative impact than transgenic lines in terms of visible symptoms (A,B), H2O2 content (C), electrolyte leakage (D), and MDA content (E). Relative expression of HvSFA2e gene in WT and transgenic lines under control and heat shock conditions. Error bars represent standard error (SE). Data represent mean ± SE. Asterisks (*, **, ***) were used to indicate significant differences between WT and transgenic lines calculated using Student’s t test, *P value < 0.05, **P value < 0.01, and ***P value < 0.001
The measurement of MDA content and electrolyte leakage showed the same trend as there was no significant difference among the WT and transgenic plants under control condition, though under heat stress, WT plants showed significantly high MDA content (3.12 µmol/g FW) and electrolyte leakage (38.88%) in comparison to transgenic lines (MDA content—TR4: 1.44, TR5: 1.50 µmol/g FW; electrolyte leakage—TR4: 26.82%, TR5: 27.89%) (Fig. 4D, E). These results show that the HvHSFA2e overexpression lines are comparatively better in maintaining the membrane integrity and limiting the lipid peroxidation than WT plants under heat stress conditions.
Overexpression of HvHSFA2e improves the drought tolerance in transgenic barley lines
To analyze the impact of HvHSFA2e overexpression on plant drought tolerance, WT and transgenic plants were grown up to 2 weeks under optimal controlled condition, then subjected to drought condition by stopping the watering for next 2 weeks. As shown in Fig. 5, severe drought-induced leaf wilting was observed in WT plants after a drought treatment of 2 weeks, although the symptoms were less severe in case of transgenic plants. Transgenic lines maintained higher RWC and low electrolyte leakage, whereas WT plants showed loss in RWC and increase in electrolyte leakage under stress conditions. These findings indicate the role HvHSFA2e in enhancement of water retention capacity and cell membrane stability under water-limiting condition for improved plant performance.

Response of wild-type (WT) and HvHSFA2e overexpression lines under drought stress. 2-week-old WT and overexpression lines were subjected to drought stress by stopping the watering for next 2 weeks. WT plants started showing early leaf wilting in comparison to transgenic lines (A, B), and transgenic lines maintained RWC under water-limiting condition (C), H2O2 content (D), electrolyte leakage (E), and MDA content (F), relative expression of HvSFA2e gene in WT and transgenic lines under control and drought stress conditions (G). Data represent mean ± SE. Asterisks (*, **, ***) were used to indicate significant differences between WT and transgenic lines calculated using Student’s t test, *P value < 0.05, **P value < 0.01, and ***P value < 0.001
Global transcriptome analysis to reveal the HvHSFA2e-mediated modulation in major stress-related transcriptional cascades
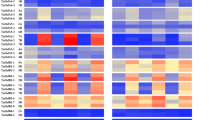
The HSFs of subclass A2 have been reported for their role in transcriptional regulation of the genes related to heat stress response (HSR) (Ogawa et al. 2007; Xin et al. 2017; Li et al. 2018). RNA-seq analysis of overexpression lines TR4, TR5, and wild-type barley plants WT1, WT2 revealed the modulations in transcriptome (Fig. 6A).


A Heat maps showing modulations in transcriptome of HvHSFA2e overexpression lines in comparison to WT control. Red color represents upregulation and green color shows down regulation, the color legend depicts the expression levels. B Illustration of GO terms enrichment for DEGs utilizing the BiNGO plugin within Cytoscape and visualized via the REVIGO web server. Subsections A–B denote biological process terms (both upregulated and downregulated), sections C–D depict cellular component terms (both upregulated and downregulated), and segments E–F showcase molecular function terms (both upregulated and downregulated). Enriched significant GO terms are represented using corrected P values (color scale) and log values (bubble size). (DEGs: differentially expressed genes
A total of 893 genes were differentially expressed in HvHSFA2e overexpression lines, with 584 upregulated and 309 downregulated. Gene Ontology (GO) enrichment analysis identified the functional distribution of these transcripts. The enriched GO terms, visualized using REVIGO (Fig. 6B), were categorized into biological processes (stress response, thermotolerance, transport, development, macromolecule biosynthesis, and photosystem), molecular functions (nucleic acid binding, transferase activity, oxidoreductase activity, terpene synthesis, vacuolar traffic, metal ion binding, and transmembrane transport), and cellular components (plasma membrane, nucleus, chloroplast, mitochondria, Golgi apparatus, ubiquitin ligase complex).
Major differentially expressed genes (DEGs) included heat shock factors (HSFs), heat shock proteins (HSPs), ABC transporters, antioxidant enzymes, Ca2+-mediated signaling, and genes related to terpene and flavonoid biosynthesis (Fig. 6A). Specifically, ten HSF genes and various HSP family genes (HvHSP20, HvHSP70, HvHSP90, HvHSP100), along with molecular chaperones and chaperonins involved in protein homeostasis and stress tolerance, were upregulated.
Antioxidant enzyme genes like HvMDAR, peroxidase, HvGSHB, HvGSTF, and HvGPX showed increased expression, helping to mitigate ROS accumulation under stress. Ca2+-mediated signaling genes, including calcium-binding proteins, CIPK19, calcium uniporter protein, calcium sensing receptor, and CBPK, were also upregulated, highlighting their role in stress tolerance.
In addition, the overexpression of HvHSFA2e resulted in upregulation of ABA-related genes (NDR1/HIN1-like proteins, NCED3, bHLH transcription factors), terpene biosynthesis genes (HvTPS1, HvTPS10, HvLIS, E-beta-caryophyllene synthase, zeta-carotene desaturase), and flavonoid biosynthesis genes (HvCHS2, HvCHI3, WD repeat-containing protein, flavonoid O-methyltransferase, flavanone 3-dioxygenase 2, flavonoid 3'-monooxygenase).
These findings suggest that HvHSFA2e overexpression influences a wide range of stress response mechanisms, involving the secondary metabolism and regulatory pathways in barley.
Validation of differential expression of HSP and HSF genes through qRT-PCR
The HSP and HSF family genes which play major role in HSR and thermotolerance showed differential expression in transcriptome analysis. The validation of their differential expression was done using qRT-PCR; the genes selected for qRT-PCR validation were HvHSP70-2, HvHSP90-1, HvHSP100-2, HvHSP100-3, HvHSFA6b, and HvHSFB2c. The result of qRT-PCR experiment showed consistency with RNA-seq data (Fig. 7), confirming the differential expression of these genes in HVHSFA2e lines as well as good quality of transcriptome data.

Validation of differential gene expression in RNA-seq data through qRT-PCR analysis. qRT-PCR analysis of some upregulated abiotic stress-responsive genes in RNA-seq data in transgenic lines (TR4-1, TR5-1, and TR8-1) and wild-type (WT) plants. y-axis represent the relative fold change in gene expression level, snoR14 gene was used as internal control reference for calculating the expression by the 2^−ddCt method. Data represent mean ± SE. Asterisks (*, **, ***) were used to indicate significant differences between WT and transgenic lines calculated using Student’s t test, *P value < 0.05, **P value < 0.01, and ***P value < 0.001
Discussion
HvHSFA2e is a heat-inducible transcription regulator
The HSF genes from model plants have been characterized for their role in plant heat stress response; however, there is a scarcity of information about the role of these genes in stress response of cereal crops. Investigating the HSF gene family in barley (Mishra et al. 2020), we realized that these genes may contribute to the enhancement of multi-stress tolerance in crop plants. To evaluate this hypothesis, we cloned HvHSFA2e, a heat and drought inducible gene, to functionally characterize it through overexpression barley plants. The subcellular localization study revealed the nuclear-cytoplasmic nature of HvHSFA2e protein; the presence of HvHSFA2e protein in the nucleus suggests its transactivation activity and regulatory role. The localization of FvHSFA2a-GFP and AtHSFA6b-YFP proteins showed a similar trend as within the plant cell, these were also distributed among nucleus and cytosol (Hu et al. 2015; Huang et al. 2016).
In wild-type plants, the expression of the HvHSFA2e gene is inducible in nature; hence for the functional characterization, we cloned full-length CDNA of this gene under the regulation of a constitutive promoter (ZmUbi) for overexpression. The ZmUbi promoter has been well studied and employed to achieve the constitutive expression of the targeted genes in cereal crops such as wheat, rice, and maize (Zang et al. 2018; Achary et al. 2020; Li et al. 2022a). The genetic transformation of barley cv. Golden promise was performed using the binary vector pANIC6B (Mann et al. 2012). A. tumefaciens does not naturally infect the monocot plants, yet the Agrobacterium-mediated genetic transformation has been successfully used to develop the stable transgenics in many monocotyledonous species including barley, rice, wheat, maize, and triticale (Ziemienowicz et al. 2012; Zhang et al. 2016; Char et al. 2017; Hayta et al. 2019; Poonia et al. 2020). In our study, we used immature embryos of barley as explant and generated stable transgenic lines overexpressing HvHSFA2e gene through Agrobacterium-mediated genetic transformation approach. The transgenic lines showed stable constitutive expression of HvHSFA2e, which is otherwise inducible in wild-type plants. To investigate the transcription regulatory role of HvHSFA2e gene in thermotolerance as well as other abiotic stress responses, the impact of HvHSFA2e overexpression on barley global transcriptome was accessed through RNA-seq analysis.
The HSF genes are the key transcriptional regulators under adverse environmental conditions; not only in heat but also under other abiotic stresses, the HSFs modulate the transcriptional cascades very precisely to confer stress tolerance in plant (Nover et al. 2001; Andrasi et al. 2021). The transactivation assay revealed that HvHSFA2e possesses the transcriptional activation potential and may regulate the transcription of multiple downstream genes to modulate the stress response. In transcriptome analysis, several abiotic stress response-related genes were shown to be positively regulated in barley HvHSFA2e overexpression lines. Earlier, the overexpression studies showed the similar results for different HSF genes belonging to subclass A2 (Ogawa et al. 2007; Nishizawa-Yokoi et al. 2009; Banti et al. 2010; Li et al. 2024). In our study, we found that most of the DEGs were involved in heat shock response, drought stress response, Ca2+-mediated signaling pathway, ABA biosynthesis and signaling pathway, ethylene biosynthesis, flavonoid and terpene metabolism.
HvHSFA2e is a key regulator of Ca2+-mediated abiotic stress signaling and response pathways in barley
Ca2+ acts as a macronutrient and secondary messenger in plant growth, development, and stress response processes. It has a significant role in maintaining the membrane stability, hormonal regulation, and enzymatic reactions (Ahmad et al. 2016). The upregulation of calcium sensing receptor (CAS), calcium uniporter protein, calcium-dependent protein kinase 10 (CDPK10), Calmodulin, calcium-binding protein PBP1, and calcyclin-binding protein (CYBP) was found in HvHSFA2e overexpression lines. The CDPKs are involved in phosphorylation-based activation of stress-responsive proteins. Zhao et al. 2021 reported that maize CDPK7 interacts and phosphorylates a HSP17 protein under high temperature stress. In the same study, they found that maize transgenics overexpressing ZmCDPK7 showed enhanced thermotolerance in terms of reduced ROS content and improved concentration of antioxidants. We also noted the upregulation of Respiratory burst oxidase homolog (RBOH) gene which encodes a Ca2+-dependent NADPH oxidase. The NADPH oxidases generate the ROS under adverse environmental conditions to initiate the ROS-dependent stress response mechanism in plants (Wang et al. 2018b; Navathe et al. 2019). Sun et al. 2019 reported that RBOH-generated superoxide is necessary to maintain the heat stress memory, and strong suppression of RBOH resulted in reduced acquired thermotolerance in tomato plants. Several studies based on identification and expression analysis of RBOH genes in crop and model plants depicted their essential role in ROS-dependent signaling and abiotic stress tolerance (Torres and Dangl 2005; Miller et al. 2008; Sun et al. 2019).
Most of the class A HSF genes possess transactivation activity and activate the expression of other HSFs, HSPs, and antioxidative defense system-related genes (Xin et al. 2010; Lavania et al. 2018; Singh et al. 2021; Li et al. 2022b). The HSFA2a, A2c, A2d, A2e, A2f, A3, and A7 genes showed significant upregulation in rice plants under heat stress conditions (Chauhan et al. 2011). Out of 23 barley HSFs, a total of 10 genes, namely HvHSFA2a, HvHSFA2e, HvHSFA4a, HvHSFA6a, HvHSFA6b, HvHSFA7b, HvHSFB1, HvHSFB2b, HvHSFB2c, and HvHSFC2b showed upregulation in barley HvHSFA2e overexpression lines. These observations suggest that along with constitutively expressing HvHSFA1a, heat-inducible HvHSFA2e acts as a key modulator of transcriptional cascades under high-temperature stress. These upregulated HSFs have been reported to improve abiotic stress tolerance when overexpressed in plants. Meena et al. (2022) reported that TaHSFA6b-D is localized to nucleus under heat stress and links the unfolded protein response with HSR to regulate the protein homeostasis under stress conditions. Overexpression of LlHSFA4 improves the basic thermotolerance through the modulation in expression of downstream genes of HSR (Wang et al. 2022). The higher expression level of HSF genes has been reported to upregulate the HSP gene of different classes. Poonia et al. (2020) reported the upregulation of Small HSPs, HSP70, HSP90, and chaperonin60 genes in barley transgenic lines overexpressing a wheat heat stress transcription factor, TaHSFA6b. The HSPs have been well characterized for their role in structural and conformational maintenance of heat susceptible proteins. Most of the overexpression studies of HSF genes have reported the constitutive upregulation of HSP genes. Xin et al. 2010 found that HSP25, HSP70, and HSP101 genes expressed constitutively in Arabidopsis transgenic plants overexpressing the lily HSFA2 gene. Recently, Li et al. (2024) also reported that overexpression of TaHSfA2-11 upregulated many heat response genes, including HSP16.6 and HSP21. Similarly, we found the upregulation of HvHSP20, HvHSP70, HvHSP90, and HvHSP100 genes in barely HvHSFA2e overexpression lines. Among the HSP90 genes, HvHSP90-4 and HvHSP90-5 showed significant upregulation in our study. The HSP90 gene from soybean ameliorates the negative impacts of abiotic stress when overexpressed in Arabidopsis plants (Xu et al. 2013). HSP100/ClpB proteins show environmental conditions and development-specific expression in plants and devoid of these proteins make the plants extremely sensitive to heat stress (Mishra and Grover 2016). The four HSP100 genes (HvHSP100-1, HvHSP100-2, HvHSP100-3 and HvHSP100-4) showed an increase ranging from 8- to 11-folds in their expression in barely transgenic lines in present study. Keeler et al. (2000) reported that HSP100/ClpB proteins contribute to acquire thermotolerance in lima bean plants, based on their HSR-associated expression patterns. HSP70 proteins are essential for maintaining the native conformation of heat sensitive proteins; as soon as heat shock strikes, these proteins dissociate from the HSF/HSP complex to bind with heat-labile proteins, releasing the HSFs for activation of HSR (Sung et al. 2001). We observed the upregulation of HvHSP70-2, HvHSP70-4, HvHSP70-5 genes in barley transgenic lines. The higher expression levels of HSP70 genes have been reported to contribute in thermotolerance enhancement; such as BcHSP70, showing the maximum similarity with AtHSP70-4 gene and Paeonia lactiflora HSP70 conferred the thermotolerance when overexpressed in tobacco and Arabidopsis plants, respectively (Wang et al. 2016; Zhao et al. 2019).
In addition to HSPs, antioxidative defense systems, including the enzymatic and non-enzymatic antioxidants, play a pivotal role in protecting the molecular and physiological processes which are jeopardized by oxidative damage caused by heat stress in plants (Caverzan et al. 2016). Glutathione (GSH) and ascorbate are two key regulators of a cell’s redox homeostasis. In Halliwell–Asada pathway (Ascorbate-Glutathione cycle), the GSH-mediated recycling of ascorbate occurs and the enzymes involved are ascorbate peroxidase (APX), monodehydroascorbate reductase (MDHAR), dehydroascorbate reductase (DHAR), and glutathione reductase (GR). GSH is one of the major thiol antioxidants found in plants, the capability interchange in reduced and oxidized forms enables the glutathione to maintain the redox homeostasis (Noctor et al. 2011). GSH-mediated redox homeostasis and glutathione biosynthesis-related genes—Glutathione synthetase (GSH2), Glutathione peroxidases (GPX4 and GPX6) and Glutathione S transferases (GSTF1, GSTT3, and GSTU6)—notably upregulated in HvHSFA2e overexpression lines. GSH2 gene catalyzed the biosynthesis of glutathione in plants and GPX genes mediate the glutathione-dependent scavenging of peroxide species to protect the plant cell from oxidative damage (Park et al. 2017; Li et al. 2021; Madhu et al. 2023). GST genes have been reported to express differentially under abiotic stress conditions as well as the enzyme encoded by these genes facilitate the glutathione conjugation to reduce the ROS accumulation (Liang et al. 2018; Kumar et al. 2018). The GPX genes have been reported to differentially express under stress conditions, the GPX6 strongly upregulated in stress-exposed Arabidopsis plants and Filiz et al. (2019) suggested this might be the source of mitochondrial ROS generation to initiate the stress response pathways.
Ascorbate is an important ROS eliminating member of the plant antioxidant defense system as it can directly convert peroxide species into water molecules (Arora et al. 2002). The enzymes which regulate the cellular ascorbate pool are APX, MDHAR, and DHAR. We found upregulation of MDHAR1 gene, encoding the enzyme monodehydroascorbate reductase 1 in HvHSFA2e overexpression lines. MDHAR enzyme catalyzes the conversion of monodehydroascorbate to ascorbic acid and the increased expression of corresponding gene has been observed to enhance the abiotic stress tolerance in plants (Yeh et al. 2019; Tanaka et al. 2021).
Role of transcription factors in HvHSFA2e-mediated abiotic stress response
Transcription factors determine the transcriptome by regulating the gene expression in a condition specific manner (Chen and Zhu 2004). To survive in adverse environment, highly specific and precisely programed modulations take place in transcription profiles in plants. The RNA-seq analysis of WT and HvHSFA2e overexpression lines showed the differential expression of HSFs, WRKY, MYB, MBF1C (multiprotein bridging factor 1c), bZIP (basic region/leucine zipper motif), ERF (ethylene-responsive factor), and NAC transcription factors, which are the major transcriptional regulators participating in reprograming transcriptome during abiotic stress response. Among the HSFs, mainly the class A genes were upregulated which are known for their role in thermotolerance (Xin et al. 2010). WRKY transcription factors are exclusive to the plant kingdom; these are involved in plant growth, development, and response to environmental stress. The WRKY gene family members, WRKY19, WRKY28, and WRKY71, showed upregulation in present study. Arabidopsis WRKY25 and 26 upregulate the transcription of thermotolerance-related genes including HSPs and ZATs (Li et al. 2009, 2011). Contrary to WRKY genes, MYB genes are present in all the eukaryotic genomes. MYB20 and MYB59 genes showed upregulated transcription in HvHSFA2e overexpression lines. MYB20-overexpressing Arabidopsis plants showed enhanced salt tolerance (Cui et al. 2013), whereas overexpression of rice MYB55 gene in maize upregulated the stress-related genes, consequently enhancing the performance of transgenics under high-temperature stress (Casaretto et al. 2016).
The MBF1 genes show differential expression under abiotic and biotic stresses. The MBF1C gene was highly upregulated in HvHSFA2e overexpression lines in comparison to WT plants. The heat stress exposure has been reported to induce the accumulation of MBF1C protein leaf tissue; the mbf1c mutants showed a decrease in basal thermotolerance. Though, MBF1C overexpressing Arabidopsis transgenics showed increased basal and acquired thermotolerance in comparison to wild-type plants (Suzuki et al. 2008). In Arabidopsis triple mbf1abc, the seedlings showed the upregulation of HSP70 and when this mutation was complemented with MBF1C, the expression level of HSP70 was found similar to wild-type plants, showing that MBF1C is a negative regulator of HSP70 (Arce et al. 2010). Recently, Tian et al. (2022) showed that wheat MBF1C regulates the translation of heat HSR-specific genes and it localizes along with stress granules.
These observations show that the upregulation of these transcription factors in HvHSFA2e overexpression lines play major role in transcriptional modifications which improves the abiotic stress response of transgenic lines.
HvHSFA2e overexpression modulates the ABA and flavonoid biosynthetic pathways in barley
The phytohormones play an important role in various signaling pathways. The involvement of abscisic acid (ABA) has been reported in acquired abiotic stress tolerance of plants. The modulation in endogenous ABA concentration determines the expression of several stress-responsive genes. The abi-1 mutants, which are ABA insensitive due to the mutated ABA sensing protein phosphatase (ABA insensitive), showed increased heat sensitivity in comparison to wild-type plants; the abi-1 mutants showed retarded growth and increased mortality at 37 °C (Larkindale and Knight 2002). Three major genes identified for their role in ABA biosynthesis are 9-cis-epoxycarotenoid dioxygenase (NCED3), zeaxanthin epoxidase (ZEP), and abscisic aldehyde oxidase (AAO) (Seo et al. 2000; Iuchi et al. 2001). The wild-type barley plants showed upregulation of HvHSFA2e gene in vegetative root and shoot tissues when treated with ABA (Mishra et al. 2020). In present study, the HvHSFA2e overexpression lines showed the constitutive upregulation of zeta-carotene desaturase (ZDS), NCED1, NCED3, AAO, NDR1/HIN1-like proteins, and bHLH transcription factors which are key genes for ABA biosynthesis and signaling. Zeta-carotene desaturase (ZDS) is an essential enzyme for carotenoid biosynthesis which catalyzes the desaturation of zeta-carotene and forms neurosporene. The zds mutant showed hampered carotenoid biosynthesis (Dong et al. 2007). Carotenoids not only work as antioxidants but also act as a precursor for ABA biosynthesis in plants; thus, the impaired carotenoid biosynthesis may lead to a lack of endogenous ABA levels. The constitutive expression of AgZDS gene resulted in the increased level of carotenoids in transgenic Arabidopsis plants (Ding et al. 2021). The NCED gene encodes 9-cis-epoxycarotenoid dioxygenase enzyme which catalyzes the oxidative cleavage in a rate-limiting step of ABA biosynthesis. The RiNCED1 gene in raspberry showed induction under heat, cold, salt stress as well as upon ABA treatment. The higher ABA content and improved stress tolerance was observed in the transgenic Arabidopsis plants overexpressing the RiNCED gene (Yang et al. 2021). Similarly, the ectopic expression of the rice NCED3 gene in Arabidopsis conferred drought tolerance through increased ABA content and reduction in water loss (Hwang et al. 2010). The multiple aldehyde oxidase (ALDO) genes are involved in ABA biosynthesis in plants; among these genes, the AAO3/ALDO3 gene has major contribution in maintaining the endogenous ABA levels (Gonzalez-Guzman et al. 2004). Other than ABA biosynthesis-related genes, we observed the upregulation of NDR1/HIN1-like protein 6 in HvHSFA2e overexpression lines. NDR1/HIN1-like genes show strong induction under ABA treatment as well as these genes assist the ABA-signaling mediated abiotic stress responses in plants (Bao et al. 2016).
Ethylene is known as fruit ripening and senescence-related plant hormone; however, it also has an important role in plant stress response (Iqbal et al. 2017). The three enzymes, S-adenosyl-l-methionine synthetase (SAM synthetase), 1-aminocyclopropane-1-carboxylic synthase (ACC synthase), and 1-aminocyclopropane-1-carboxylate oxidase (ACC oxidase), regulate the ethylene biosynthesis process. ACO gene-encoded ACC oxidase is exclusively dedicated to catalyze the final step of ethylene biosynthesis from S-adenosyl-l-methionine (Ruduś et al. 2013; Pattyn et al. 2021). In our study, HvHSFA2e overexpression lines showed significantly high expression of ACO, ERF109, ERF54, ERF60 genes in comparison to wild-type barley plants. Recently, Huang et al. (2021) reported that ethylene response factors (ERFs) may directly enhance the thermotolerance by regulating the transcription of HSFA2 gene and subsequently increasing the expression of other abiotic stress-responsive genes. In the same study, they found that the mutation in multiple ERF genes results in compromised basal thermotolerance.
The plant secondary metabolism is greatly influenced by the environmental cues. Metabolic reprogramming is one of the acclimation methods in plants to survive the environmental stress. The secondary metabolites have diverse physiological functions such as ROS homeostasis, protection from photo inhibition, enzyme activation, regulation of osmolarity and signal regulation (Zandalinas et al. 2017; Delfin et al. 2019). Flavonoids are present across the plant kingdom and these secondary metabolites have been studied for their antioxidant properties. Under the environmental stresses, the increase in flavonoid biosynthesis and accumulation has been reported in vegetative plant tissues (Winkel-Shirley 2001, 2002). The chalcone synthase (CHS) and chalcone isomerase (CHI) are the genes responsible for flavonoid biosynthesis regulated by Myb/bHLH transcription factors (Xu et al. 2015). The transcription of CHS2, CHI3, bHLH (bHLH6, bHLH148, bHLH162, bHLH167 and bHLH168), and Myb (Myb20 and Myb59) genes was higher in HSFA2e overexpression lines in comparison to wild-type barley plants. Many previous and recent studies have reported the stress inducible nature of CHS genes; moreover, the characterization of an okra CHS gene in Arabidopsis revealed its role in enhancing the abiotic stress tolerance (Wang et al. 2018a, 2022; Yao et al. 2019; Singh and Kumaria 2020).
Terpenes are the volatile secondary metabolites which enable the plants to interact directly with biotic and abiotic both kinds of environmental factors. A wide and diverse range of terpenes is generated from geranyl diphosphate (GPP) and farnesyl diphosphate (FPP) precursors by the activity of terpene synthase enzymes, encoded by TPS gene. In HvHSFA2e overexpression lines, the upregulation of TPS1 (acyclic sesquiterpene synthase), TPS23 (beta-caryophyllene synthase), LIS (linalool synthase) genes was observed. In previous studies, the differential expression of TPS genes has been reported under high temperature and other abiotic stress conditions (Zhang et al. 2019). Lee et al. (2015) reported the conversion of GPP into limonene and other monoterpenes by rice TPS20 protein; moreover, when subjected to abiotic stress, the rice TPS20 gene showed an upregulated transcription. Based on their experiments on Quercus seedlings, Peñuelas and Llusià (2002) suggested that monoterpenes may contribute to thermotolerance by increasing the photochemical efficiency and reducing the photodamage at high temperatures.
Conclusion
Functional characterization of stress response regulating genes in crop plants is essential to mitigate the yield reductions caused by heat stress. In this study, we characterized HvHSFA2e gene, which exhibited the highest expression in the vegetative shoot of wild-type barley exposed to a 30-min heat shock. Transcriptome analysis of transgenic lines revealed differential expression of genes related to plant stress response and ABA biosynthesis. The findings of present study suggests that HvHSFA2e plays a key role as transcriptional regulator in plant stress responses (Fig. 8). Biochemical analysis showed increased activation of enzymatic antioxidants and a subsequent reduction in ROS accumulation, contributing to enhanced drought and heat tolerance in the transgenic lines. Based on these findings, we propose that the HvHSFA2e gene is a promising target for genetic manipulation to develop the stress-tolerant varieties in temperate cereals to sustain the adverse environmental conditions such as high temperature and water depletion.

Schematic representation displaying the role of HvHSFA2e gene in abiotic stress response and tolerance. HvHSFA2e overexpression upregulated the expression of RbohD gene which is responsible for the initiation of ROS-mediated stress signaling. Moreover, HvHSFA2e positively regulates the expression of stress-responsive transcription factors and defense-related proteins through binding to the heat shock element (HSE) present in the promoter of these genes. These genes activate and maintain different stress mitigation pathways (including HSR) to enhance the stress tolerance in transgenic lines
Data availability
The RNA-seq data (accession number GSE241562) generated and analysed during this study is available in the GEO, NCBI database.
References
Achary VM, Sheri V, Manna M, Panditi V, Borphukan B, Ram B, Agarwal A, Fartyal D, Teotia D, Masakapalli SK, Agrawal PK (2020) Overexpression of improved EPSPS gene results in field level glyphosate tolerance and higher grain yield in rice. Plant Biotechnol J 18(12):2504–2519
Ahmad P, Abdel Latef AA, AbdAllah EF, Hashem A, Sarwat M, Anjum NA, Gucel S (2016) Calcium and potassium supplementation enhanced growth, osmolyte secondary metabolite production, and enzymatic antioxidant machinery in cadmium-exposed chickpea (Cicer arietinum L.). Front Plant Sci 7:187060
Andrási N, Pettkó-Szandtner A, Szabados L (2021) Diversity of plant heat shock factors: regulation, interactions, and functions. J Exp Bot 72(5):1558–1575
Arce DP, Godoy AV, Tsuda K, Yamazaki KI, Valle EM, Iglesias MJ, Di Mauro MF, Casalongué CA (2010) The analysis of an Arabidopsis triple knock-down mutant reveals functions for MBF1 genes under oxidative stress conditions. J Plant Physiol 167(3):194–200
Arora A, Sairam RK, Srivastava GC (2002) Oxidative stress and antioxidative system in plants. Curr Sci 82:1227–1238
Atlin GN, Cairns JE, Das B (2017) Rapid breeding and varietal replacement are critical to adaptation of cropping systems in the developing world to climate change. Glob Food Sec 12:31–37
Banti V, Mafessoni F, Loreti E, Alpi A, Perata P (2010) The heat-inducible transcription factor HsfA2 enhances anoxia tolerance in Arabidopsis. Plant Physiol 152(3):1471–1483
Bao Y, Song WM, Pan J, Jiang CM, Srivastava R, Li B, Zhu LY, Su HY, Gao XS, Liu H, Yu X (2016) Overexpression of the NDR1/HIN1-like gene NHL6 modifies seed germination in response to abscisic acid and abiotic stresses in Arabidopsis. PLoS ONE 11(2):e0148572
Battisti DS, Naylor RL (2009) Historical warnings of future food insecurity with unprecedented seasonal heat. Science 323(5911):240–244
Bechtold U, Albihlal WS, Lawson T, Fryer MJ, Sparrow PA, Richard F, Persad R, Bowden L, Hickman R, Martin C, Beynon JL (2013) Arabidopsis HEAT SHOCK TRANSCRIPTION FACTORA1b overexpression enhances water productivity, resistance to drought, and infection. J Exp Bot 64(11):3467–3481
Casaretto JA, El-Kereamy A, Zeng B, Stiegelmeyer SM, Chen X, Bi YM, Rothstein SJ (2016) Expression of OsMYB55 in maize activates stress-responsive genes and enhances heat and drought tolerance. BMC Genom. https://doi.org/10.1186/s12864-016-2659-5
Caverzan A, Casassola A, Brammer SP (2016) Antioxidant responses of wheat plants under stress. Genet Mol Biol 39:1–6
Chakraborty S, Hill AL, Shirsekar G, Afzal AJ, Wang GL, Mackey D, Bonello P (2016) Quantification of hydrogen peroxide in plant tissues using amplex red. Methods 109:105–113
Char SN, Neelakandan AK, Nahampun H, Frame B, Main M, Spalding MH, Becraft PW, Meyers BC, Walbot V, Wang K, Yang B (2017) An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize. Plant Biotechnol J 15(2):257–268
Charng YY, Liu HC, Liu NY, Chi WT, Wang CN, Chang SH, Wang TT (2007) A heat-inducible transcription factor, HsfA2, is required for extension of acquired thermotolerance in Arabidopsis. Plant Physiol 143(1):251–262
Chauhan H, Khurana N, Agarwal P, Khurana P (2011) Heat shock factors in rice (Oryza sativa L.): genome-wide expression analysis during reproductive development and abiotic stress. Mol Genet Genom 286:171–187
Chen WJ, Zhu T (2004) Networks of transcription factors with roles in environmental stress response. Trends Plant Sci 9(12):591–596
Choi WG, Barker RJ, Kim SH, Swanson SJ, Gilroy S (2019) Variation in the transcriptome of different ecotypes of Arabidopsis thaliana reveals signatures of oxidative stress in plant responses to spaceflight. AJB 106(1):123–136
Cui MH, Yoo KS, Hyoung S, Nguyen HT, Kim YY, Kim HJ, Ok SH, Yoo SD, Shin JS (2013) An Arabidopsis R2R3-MYB transcription factor, AtMYB20, negatively regulates type 2C serine/threonine protein phosphatases to enhance salt tolerance. FEBS Lett 587(12):1773–1778
Delfin JC, Watanabe M, Tohge T (2019) Understanding the function and regulation of plant secondary metabolism through metabolomics approaches. Theor Exp Plant Physiol 31:127–138
Ding X, Liu JX, Li T, Duan AQ, Yin L, Wang H, Jia LL, Liu YH, Liu H, Tao JP, Xiong AS (2021) AgZDS, a gene encoding ζ-carotene desaturase, increases lutein and β-carotene contents in transgenic Arabidopsis and celery. Plant Sci 312:111043
Dionisio-Sese ML, Tobita S (1998) Antioxidant responses of rice seedlings to salinity stress. Plant Sci 135(1):1–9
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21
Dong H, Deng Y, Mu J, Lu Q, Wang Y, Xu Y, Chu C, Chong K, Lu C, Zuo J (2007) The Arabidopsis spontaneous cell death1 gene, encoding a ζ-carotene desaturase essential for carotenoid biosynthesis, is involved in chloroplast development, photoprotection and retrograde signalling. Cell Res 5:458–470
Farooq M, Bramley H, Palta JA, Siddique KH (2011) Heat stress in wheat during reproductive and grain-filling phases. Crit Rev Plant Sci 30(6):491–507
Ferdous J, Li Y, Reid N, Langridge P, Shi BJ, Tricker PJ (2015) Identification of reference genes for quantitative expression analysis of microRNAs and mRNAs in barley under various stress conditions. PLoS ONE 10(3):e118503
Filiz E, Ozyigit II, Saracoglu IA, Uras ME, Sen U, Yalcin B (2019) Abiotic stress-induced regulation of antioxidant genes in different Arabidopsis ecotypes: microarray data evaluation. Biotechnol Biotec Eq 33(1):128–143
Fridovich I (1986) Superoxide dismutases. Adv Enzymol Relat Areas Mol 58:61–97. https://doi.org/10.1002/9780470123041
Giannopolitis CN, Ries SK (1977) Superoxide dismutases: I. Occurr High Plants Plant Physiol 59:309–314
Giorno F, Wolters-Arts M, Grillo S, Scharf KD, Vriezen WH, Mariani C (2010) Developmental and heat stress-regulated expression of HsfA2 and small heat shock proteins in tomato anthers. J Exp Bot 61(2):453–462
González-Guzmán M, Abia D, Salinas J, Serrano R, Rodriguez PL (2004) Two new alleles of the abscisic aldehyde oxidase 3 gene reveal its role in abscisic acid biosynthesis in seeds. Plant Physiol 135(1):325–333
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li BO, Lieber M, MacManes MD (2013) De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nat Protoc 8:1494–1512
Hayta S, Smedley MA, Demir SU, Blundell R, Hinchliffe A, Atkinson N, Harwood WA (2019) An efficient and reproducible Agrobacterium mediated transformation method for hexaploid wheat (Triticum aestivum L.). Plant Methods 15(1):1–5
Hensel G, Kastner C, Oleszczuk S, Riechen J, Kumlehn J (2009) Agrobacterium-mediated gene transfer to cereal crop plants: current protocols for barley, wheat, triticale and maize. Int J Plant Genom 2009:835608. https://doi.org/10.1155/2009/835608
Hu Y, Han YT, Wei W, Li YJ, Zhang K, Gao YR, Zhao FL, Feng JY (2015) Identification, isolation, and expression analysis of heat shock transcription factors in the diploid woodland strawberry Fragaria vesca. Front Plant Sci 6:736
Huang YC, Niu CY, Yang CR, Jinn TL (2016) The heat stress factor HSFA6b connects ABA signaling and ABA-mediated heat responses. Plant Physiol 172(2):1182–1199
Huang J, Zhao X, Bürger M, Wang Y, Chory J (2021) Two interacting ethylene response factors regulate heat stress response. Plant Cell 33(2):338–357
Hwang SG, Chen HC, Huang WY, Chu YC, Shii CT, Cheng WH (2010) Ectopic expression of rice OsNCED3 in Arabidopsis increases ABA level and alters leaf morphology. Plant Sci 178(1):12–22
Ings J, Mur LA, Robson PR, Bosch M (2013) Physiological and growth responses to water deficit in the bioenergy crop Miscanthus giganteus. Front Plant Sci 4:468. https://doi.org/10.3389/fpls.2013.00468
Iqbal N, Khan NA, Ferrante A, Trivellini A, Francini A, Khan MI (2017) Ethylene role in plant growth, development and senescence: interaction with other phytohormones. Front Plant Sci 8:235913
Iuchi S, Kobayashi M, Taji T, Naramoto M, Seki M, Kato T, Tabata S, Kakubari Y, Yamaguchi-Shinozaki K, Shinozaki K (2001) Regulation of drought tolerance by gene manipulation of 9-cis-epoxycarotenoid dioxygenase a key enzyme in abscisic acid biosynthesis in Arabidopsis. Plant J 27(4):325–333
Keeler SJ, Boettger CM, Haynes JG, Kuches KA, Johnson MM, Thureen DL, Keeler CL Jr, Kitto SL (2000) Acquired thermotolerance and expression of the HSP100/ClpB genes of lima bean. Plant Physiol 123(3):1121–1132
Kumar S, Trivedi PK (2018) Glutathione S-transferases: role in combating abiotic stresses including arsenic detoxification in plants. Front Plant Sci 9:364396
Lamaoui M, Jemo M, Datla R, Bekkaoui F (2018) Heat and drought stresses in crops and approaches for their mitigation. Front Chem 6:26. https://doi.org/10.3389/fchem.2018.00026
Lämke J, Brzezinka K, Bäurle I (2016) HSFA2 orchestrates transcriptional dynamics after heat stress in Arabidopsis thaliana. Transcription 7(4):111–114
Larkindale J, Knight MR (2002) Protection against heat stress-induced oxidative damage in arabidopsis involves calcium, abscisic acid, ethylene, and salicylic acid. Plant Physiol 128(2):682–695
Lavania D, Dhingra A, Grover A (2018) Analysis of transactivation potential of rice (Oryza sativa L.) heat shock factors. Planta 247:1267–1276
Lee GW, Lee S, Chung MS, Jeong YS, Chung BY (2015) Rice terpene synthase 20 (OsTPS20) plays an important role in producing terpene volatiles in response to abiotic stresses. Protoplasma 252:997–1007
Li X (2011) Infiltration of Nicotiana benthamiana protocol for transient expression via Agrobacterium. Bio-Protoc. https://doi.org/10.21769/BioProtoc.95
Li B, Dewey CN (2011) RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform 12:1–6
Li C, Chen Q, Gao X, Qi B, Chen N, Xu S, Chen J, Wang X (2005) AtHsfA2 modulates expression of stress responsive genes and enhances tolerance to heat and oxidative stress in Arabidopsis. Sci China Ser C: Life Sci 48:540–550
Li S, Fu Q, Huang W, Yu D (2009) Functional analysis of an Arabidopsis transcription factor WRKY25 in heat stress. Plant Cell Rep 28:683–693
Li S, Fu Q, Chen L, Huang W, Yu D (2011) Arabidopsis thaliana WRKY25 WRKY26 and WRKY33 coordinate induction of plant thermotolerance. Planta 233:1237–1252
Li PS, Yu TF, He GH, Chen M, Zhou YB, Chai SC, Xu ZS, Ma YZ (2014) Genome-wide analysis of the Hsf family in soybean and functional identification of GmHsf-34 involvement in drought and heat stresses. BMC Genom 15:1–6
Li JY, Li D, Du X, Li H, Wang D, Xing Q, Yao R, Sun MY, Shi L (2018) Modular organization analysis of specific naringin/neoeriocitrin related gene expression induced by UVC irradiation in Drynaria roosii. Environ Exp Bot 156:298–315
Li W, Huai X, Li P, Raza A, Mubarik MS, Habib M, Fiaz S, Zhang B, Pan J, Khan RS (2021) Genome-wide characterization of glutathione peroxidase (GPX) gene family in rapeseed (Brassica napus L.) revealed their role in multiple abiotic stress response and hormone signaling. Antioxidants 10(9):1481
Li HG, Yang Y, Liu M, Zhu Y, Wang HL, Feng CH, Niu MX, Liu C, Yin W, Xia X (2022a) The in vivo performance of a heat shock transcription factor from populus euphratica, PeHSFA2, promises a prospective strategy to alleviate heat stress damage in poplar. Environ Exp Bot 201:104940
Li XD, Gao YQ, Wu WH, Chen LM, Wang Y (2022b) Two calcium-dependent protein kinases enhance maize drought tolerance by activating anion channel ZmSLAC1 in guard cells. Plant Biotechnol J 20(1):143–157
Li G, Liu Z, Zhang H, Zhao B, Zhang Y, Ma Z, Duan S, Meng X, Guo X (2024) Molecular characterization of a novel heat shock transcription factor gene TaHsfA2-11 and its overexpression improves thermotolerance in wheat. Environ Exp Bot 218(218):105609
Liang D, Gao F, Ni Z, Lin L, Deng Q, Tang Y, Wang X, Luo X, Xia H (2018) Melatonin improves heat tolerance in kiwifruit seedlings through promoting antioxidant enzymatic activity and glutathione S-transferase transcription. Molecules 23(3):584
Lichtenthaler HK, Wellburn AR (1983) Determinations of total carotenoids and chlorophylls a and b of leaf extracts in different solvents. Biochem Soc Trans 11(5):591–592. https://doi.org/10.1042/bst0110591
Liu Z, Friesen T (2012) DAB staining and visualization of hydrogen peroxide in wheat leaves. Bio-Protoc. https://doi.org/10.21769/BioProtoc.309
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25(4):402–408
Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Bio 15:1–21
Madhu SA, Kaur A, Tyagi S, Upadhyay SK (2023) Glutathione peroxidases in plants: innumerable role in abiotic stress tolerance and plant development. JPGR 42(2):598–613
Maere S, Heymans K, Kuiper M (2005) BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21(16):3448–3449
Mann DG, LaFayette PR, Abercrombie LL, King ZR, Mazarei M, Halter MC, Poovaiah CR, Baxter H, Shen H, Dixon RA, Parrott WA (2012) Gateway-compatible vectors for high-throughput gene functional analysis in switchgrass (Panicum virgatum L.) and other monocot species. Plant Biotechnol J 10(2):226–236
Masson-Delmotte V, Zhai P, Pörtner HO, Roberts D, Skea J, Shukla PR, Pirani A, Moufouma-Okia W, Péan C, Pidcock R (2019) Connors S (2019) Global warming of 1.5 C. An IPCC Special Report on the Impacts of Global Warming of 1:93–174
Mckersie BD, Leshem YA, Mckersie BD, Leshem YA (1994) Chilling stress. Stress and stress coping in cultivated plants. Springer, Dordrecht, pp 79–103
Meena S, Samtani H, Khurana P (2022) Elucidating the functional role of heat stress transcription factor A6b (TaHsfA6b) in linking heat stress response and the unfolded protein response in wheat. Plant Mol Biol 108:621–634
Miller G, Shulaev V, Mittler R (2008) Reactive oxygen signaling and abiotic stress. Physiol Plant 133(3):481–489
Mishra RC, Grover A (2016) Clp/BHsp100 proteins and heat stress tolerance in plants. Crit Rev Biotechnol 36(5):862–874
Mishra SK, Poonia AK, Chaudhary R, Baranwal VK, Arora D, Kumar R, Chauhan H (2020) Genome-wide identification, phylogeny and expression analysis of HSF gene family in barley during abiotic stress response and reproductive development. Plant Gene 23:100231
Mittler R, Zandalinas SI, Fichman Y, Van Breusegem F (2022) Reactive oxygen species signalling in plant stress responses. Nat Rev Mol Cell Biol 23(10):663–679
Nakano Y, Asada K (1981) Hydrogen peroxide is scavenged by ascorbate-specific peroxidase in spinach chloroplasts. Plant Cell Physiol 22:867–880. https://doi.org/10.1093/oxfordjournals.pcp.a076232
Navathe S, Singh S, Singh VK, Chand R, Mishra VK, Joshi AK (2019) Genome-wide mining of respiratory burst homologs and its expression in response to biotic and abiotic stresses in Triticum aestivum. Genes Genom 41:1027–1043
Nelson GC, Rosegrant MW, Palazzo A, Gray I, Ingersoll C, Robertson RD, Tokgoz S, Sulser TB, Ringler C, Msangi S, You L (2010) Food security, farming, and climate change to 2050: scenarios, results, policy options, vol 172. International Food Policy Research Institute (IFPRI)
Neuwirth E (2014) RColorBrewer: colorbrewer palettes. R package version1. 1–2. http://CRAN.R-project.org/package=RColorBrewer. Accessed 25 Dec 2021
Nishizawa-Yokoi A, Yoshida E, Yabuta Y, Shigeoka S (2009) Analysis of the regulation of target genes by an Arabidopsis heat shock transcription factor, HsfA2. Biosci Biotechnol Biochem 73(4):890–895
Noctor G, Queval G, Mhamdi A, Chaouch S, Foyer CH (2011) Glutathione. The Arabidopsis Book/am Soc Plant Biol 9(1):1–32
Nover L, Scharf KD, Gagliardi D, Vergne P, Czarnecka-Verner E, Gurley WB (1996) The Hsf world: classification and properties of plant heat stress transcription factors. Cell Stress Chaperones 1(4):215
Nover L, Bharti K, Döring P, Mishra SK, Ganguli A, Scharf KD (2001) Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperones 3:177
Ogawa D, Yamaguchi K, Nishiuchi T (2007) High-level overexpression of the Arabidopsis HsfA2 gene confers not only increased themotolerance but also salt/osmotic stress tolerance and enhanced callus growth. J Exp Bot 58(12):3373–3383
Park SI, Kim YS, Kim JJ, Mok JE, Kim YH, Park HM, Kim IS, Yoon HS (2017) Improved stress tolerance and productivity in transgenic rice plants constitutively expressing the Oryza sativa glutathione synthetase OsGS under paddy field conditions. J Plant Physiol 215:39–47
Patel RK, Jain M (2012) NGS QC Toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7(2):e30619
Pattyn J, Vaughan-Hirsch J, Van de Poel B (2021) The regulation of ethylene biosynthesis: a complex multilevel control circuitry. New Phytol 229(2):770–782
Peñuelas J, Llusià J (2002) Linking photorespiration, monoterpenes and thermotolerance in Quercus. New Phytol 155(2):227–237
Pérez-Salamó I, Papdi C, Rigó G, Zsigmond L, Vilela B, Lumbreras V, Nagy I, Horváth B, Domoki M, Darula Z, Medzihradszky K (2014) The heat shock factor A4A confers salt tolerance and is regulated by oxidative stress and the mitogen-activated protein kinases MPK3 and MPK6. Plant Physiol 165(1):319–334
Poonia AK, Mishra SK, Sirohi P, Chaudhary R, Kanwar M, Germain H, Chauhan H (2020) Overexpression of wheat transcription factor (TaHsfA6b) provides thermotolerance in barley. Planta 252:1–4
Ray DK, Gerber JS, MacDonald GK, West PC (2015) Climate variation explains a third of global crop yield variability. Nat Commun 6(1):5989
Ruduś I, Sasiak M, Kępczyński J (2013) Regulation of ethylene biosynthesis at the level of 1-aminocyclopropane-1-carboxylate oxidase (ACO) gene. Acta Physiol 35:295–307. https://doi.org/10.1007/s11738-012-1096-6
Schramm F, Ganguli A, Kiehlmann E, Englich G, Walch D, von Koskull-Döring P (2006) The heat stress transcription factor HsfA2 serves as a regulatory amplifier of a subset of genes in the heat stress response in Arabidopsis. Plant Mol Biol 60:759–772
Seo M, Koiwai H, Akaba S, Komano T, Oritani T, Kamiya Y, Koshiba T (2000) Abscisic aldehyde oxidase in leaves of Arabidopsis thaliana. Plant J 23(4):481–488
Singh N, Kumaria S (2020) Molecular cloning and characterization of chalcone synthase gene from Coelogyne ovalis Lindl. and its stress-dependent expression. Gene 762:145104
Singh G, Sarkar NK, Grover A (2021) Tango between ethylene and HSFA2 settles heat tolerance. Trends Plant Sci 26(5):429–432
Sun M, Jiang F, Zhou R, Wen J, Cui S, Wang W, Wu Z (2019) Respiratory burst oxidase homologue-dependent H2O2 is essential during heat stress memory in heat sensitive tomato. Sci Hortic 258:108777
Sung DY, Kaplan F, Guy CL (2001) Plant Hsp70 molecular chaperones: protein structure, gene family, expression and function. Physiol Plant 113(4):443–451
Supek F, Bošnjak M, Škunca N, Šmuc T (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6(7):e21800
Swindell WR, Huebner M, Weber AP (2007) Transcriptional profiling of Arabidopsis heat shock proteins and transcription factors reveals extensive overlap between heat and non-heat stress response pathways. BMC Genom 8:1–5
Tanaka M, Takahashi R, Hamada A, Terai Y, Ogawa T, Sawa Y, Ishikawa T, Maruta T (2021) Distribution and functions of monodehydroascorbate reductases in plants comprehensive reverse genetic analysis of Arabidopsis thaliana enzymes. Antioxidants 10(11):1726
Thordal-Christensen H, Zhang Z, Wei Y, Collinge DB (1997) Subcellular localization of H2O2 in plants. H2O2 accumulation in papillae and hypersensitive response during the barley—powdery mildew interaction. Plant J 6:1187–1194
Tian X, Qin Z, Zhao Y, Wen J, Lan T, Zhang L, Wang F, Qin D, Yu K, Zhao A, Hu Z (2022) Stress granule-associated TaMBF1c confers thermotolerance through regulating specific mRNA translation in wheat (Triticum aestivum). New Phytol 233(4):1719–1731
Torres MA, Dangl JL (2005) Functions of the respiratory burst oxidase in biotic interactions, abiotic stress and development. Curr Opin Plant Biol 8(4):397–403
Wang X, Yan B, Shi M, Zhou W, Zekria D, Wang H, Kai G (2016) Overexpression of a Brassica campestris HSP70 in tobacco confers enhanced tolerance to heat stress. Protoplasma 253:637–645
Wang X, Huang W, Liu J, Yang Z, Huang B (2017) Molecular regulation and physiological functions of a novel FaHsfA2c cloned from tall fescue conferring plant tolerance to heat stress. Plant Biotechnol J 15(2):237–248
Wang F, Ren G, Li F, Qi S, Xu Y, Wang B, Yang Y, Ye Y, Zhou Q, Chen (2018a) A chalcone synthase gene AeCHS from Abelmoschus esculentus regulates flavonoid accumulation and abiotic stress tolerance in transgenic Arabidopsis. Acta Physiol Plant 40:1–3
Wang W, Chen D, Zhang X, Liu D, Cheng Y, Shen F (2018b) Role of plant respiratory burst oxidase homologs in stress responses. Free Radic Res 52(8):826–839
Wang C, Zhou Y, Yang X, Zhang B, Xu F, Wang Y, Song C, Yi M, Ma N, Zhou X, He J (2022) The heat stress transcription factor LlHsfA4 enhanced basic thermotolerance through regulating ROS metabolism in lilies (Lilium longiflorum). Int J Mol Sci 23:572. https://doi.org/10.3390/ijms23010572
Warnes G. R Enhanced Heat Map—gplots package
Winkel-Shirley B (2001) Flavonoid biosynthesis. a colorful model for genetics biochemistry cell biology and biotechnology. Plant Physiol 126(2):485–493
Winkel-Shirley B (2002) Biosynthesis of flavonoids and effects of stress. Curr Opin Plant Biol 5(3):218–223
Wohlgemuth H, Mittelstrass K, Kschieschan S, Bender J, Weigel HJ, Overmyer K, Kangasjärvi J, Sandermann H, Langebartels C (2002) Activation of an oxidative burst is a general feature of sensitive plants exposed to the air pollutant ozone. Plant Cell Environ 6:717–726
Xin H, Zhang H, Chen L, Li X, Lian Q, Yuan X, Hu X, Cao L, He X, Yi M (2010) Cloning and characterization of HsfA2 from Lily (Lilium longiflorum). Plant Cell Rep 29(8):75–85
Xin H, Zhang H, Zhong X, Lian Q, Dong A, Cao L, Yi M, Cong R (2017) Over-expression of LlHsfA2b, a lily heat shock transcription factor lacking trans-activation activity in yeast, can enhance tolerance to heat and oxidative stress in transgenic Arabidopsis seedlings. Plant Cell Tissue Organ Cult 130:617–629
Xu J, Xue C, Xue D, Zhao J, Gai J, Guo N, Xing H (2013) Overexpression of GmHsp90s, a heat shock protein 90 (Hsp90) gene family cloning from soybean, decrease damage of abiotic stresses in Arabidopsis thaliana. PLoS ONE 8(7):e69810
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB–bHLH–WDR complexes. Trends Plant Sci 20(3):176–185
Xue GP, Sadat S, Drenth J, McIntyre CL (2014) The heat shock factor family from Triticum aestivum in response to heat and other major abiotic stresses and their role in regulation of heat shock protein genes. J Exp Bot 65(2):539–557
Yang G, Chen Y, Yu H, Zhang H, Han D, Guo X, Yan E, Quan H, Li T (2021) Raspberry (Rubus idaeus L.) NCED1 gene enhances high salinity and cold tolerance in Arabidopsis. In Vitro Cell Dev Biol Plant 57:811–819
Yao X, Wang T, Wang H, Liu H, Liu S, Zhao Q, Chen K, Zhang P (2019) Identification, characterization and expression analysis of the chalcone synthase family in the antarctic moss pohlia nutans. Antarct Sci 31(1):23–33
Yashavanthakumar KJ, Baviskar VS, Navathe S, Patil RM, Bagwan JH, Bankar DN, Gite VD, Gopalareddy K, Mishra CN, Mamrutha HM, Singh SK (2021) Impact of heat and drought stress on phenological development and yield in bread wheat. Plant Physiol Rep 2:357–367
Yeh HL, Lin TH, Chen CC, Cheng TX, Chang HY, Lee (2019) Monodehydroascorbate reductase plays a role in the tolerance of Chlamydomonas reinhardtii to photooxidative stress. Plant Cell Physiol 10:2167–2179
Zandalinas SI, Sales C, Beltrán J, Gómez-Cadenas A, Arbona V (2017) Activation of secondary metabolism in citrus plants is associated to sensitivity to combined drought and high temperatures. Front Plant Sci 7:232549
Zang X, Geng X, He K, Wang F, Tian X, Xin M, Yao Y, Hu Z, Ni Z, Sun Q, Peng H (2018) Overexpression of the wheat Triticum aestivum L TaPEPKR2 gene enhances heat and dehydration tolerance in both wheat and Arabidopsis. Front Plant Sci 9:1710
Zhang Y, Li J, Gao C (2016) Generation of stable transgenic rice (Oryza sativa L.) by Agrobacterium-mediated transformation. Curr Protoc Plant Biol 1(2):235–246
Zhang X, Niu M, Teixeira da Silva JA, Zhang Y, Yuan Y, Jia Y, Xiao Y, Li Y, Fang L, Zeng S, Ma G (2019) Identification and functional characterization of three new terpene synthase genes involved in chemical defense and abiotic stresses in Santalum album. BMC Plant Biol 19:1–8
Zhao D, Xia X, Su J, Wei M, Wu Y, Tao J (2019) Overexpression of herbaceous peony HSP70 confers high temperature tolerance. BMC Genom 20:1–1
Zhao Y, Du H, Wang Y, Wang H, Yang S, Li C, Chen N, Yang H, Zhang Y, Zhu Y, Yang L (2021) The calcium-dependent protein kinase ZmCDPK7 functions in heat-stress tolerance in maize. J Integr Plant Biol 63(3):510–527
Zheng YH, Li X, Li YG, Miao BH, Xu H, Simmons M, Yang XH (2012) Contrasting responses of salinity-stressed salt-tolerant and intolerant winter wheat (Triticum aestivum L.) cultivars to ozone pollution. Plant Physiol. Biochem. 52:169–78
Ziemienowicz A, Shim YS, Matsuoka A, Eudes F, Kovalchuk I (2012) A novel method of transgene delivery into triticale plants using the Agrobacterium transferred DNA-derived nano-complex. Plant Physiol 158(4):1503–1513
Acknowledgements
This work was supported by a grant (CRG-002579) to HC by SERB-DST, Government of India. SKM and AKP thank MHRD and SB, MK thank UGC for providing the research fellowships. We also thank GeneBank, NARO Japan for providing plasmid harboring Fl-cDNA of HvHSFA2e.
Funding
This study was supported by SERB INDIA,CRG-002579,Harsh Chauhan
Author information
Authors and Affiliations
Contributions
SKM raised and confirmed transgenic lines overexpressing HvHSFA2e. SKM, CC, SB, AKP, PS, MK, SG, and AK performed the experiments and analyzed data. HC, DS, and HG guided the study and finalized the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
Authors declare no conflict of interest.
Additional information
Communicated by Haitao Shi.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Mishra, S.K., Chaudhary, C., Baliyan, S. et al. Heat-stress-responsive HvHSFA2e gene regulates the heat and drought tolerance in barley through modulation of phytohormone and secondary metabolic pathways. Plant Cell Rep 43, 172 (2024). https://doi.org/10.1007/s00299-024-03251-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00299-024-03251-6