
Abstract
Key message
Protopanaxadiol (PPD) is an aglycone of dammarene-type ginsenoside and has high medicinal values. In this work, we reported the PPD production in transgenic tobacco co-overexpressing PgDDS and CYP716A47.
Abstract
PPD is an aglycone of ginsenosides produced by Panax species and has a wide range of pharmacological activities. PPD is synthesized via the hydroxylation of dammarenediol-II (DD) by CYP716A47 enzyme. Here, we established a PPD production system via cell suspension culture of transgenic tobacco co-overexpressing the genes for PgDDS and CYP716A47. The concentration of PPD in transgenic tobacco leaves was 2.3–5.7 µg/g dry weight (DW), depending on the transgenic line. Leaf segments were cultured on medium with various types of hormones to induce callus. Auxin treatment, particularly 2,4-D, strongly enhanced the production of DD (783.8 µg g−1 DW) and PPD (125.9 µg g−1 DW). Treatment with 2,4-D enhanced the transcription of the HMG-Co reductase (HMGR) and squalene epoxidase genes. PPD production reached 166.9 and 980.9 µg g−1 DW in a 250-ml shake flask culture and in 5-l airlift bioreactor culture, respectively.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The root of Panax ginseng is one of the most famous and widely used medicinal plant materials (Shibata 2001). The major pharmacologically active components of ginseng are dammarene-type ginsenosides, a diverse group of triterpenoid saponins. More than 30 types of ginsenosides have been identified. Dammarene-type ginsenosides are the major saponin constituents in Panax species (Shibata 2001) and are divided into two groups based on their aglycone structure: protopanaxadiol (PPD) and protopanaxatriol (PPT).
Ginseng roots and their products are typically administered orally. The absorption of glycosylated ginsenosides by the gastrointestinal tract is extremely low (Tawab et al. 2003). Acid or intestinal bacteria in the gastrointestinal tract degrade ginsenosides. Deglycosylated ginsenosides are more readily absorbed into the bloodstream and function as active compounds (Karikura et al. 1991). Hydrolyzed ginsenosides or ginsenoside aglycones have greater biological effects than the natural forms of ginsenosides (Hasegawa et al. 1997; Jia et al. 2004; Popovich and Kitts 2004; Bae et al. 2004). PPD, a hydrolyzed sapogenin belonging to the PPD group of ginsenosides, has more potent anticancer properties than other types of ginsenosides (Jia et al. 2004; Popovich and Kitts 2004; Qi et al. 2010). PPD produces apoptotic effects in cancer cells through various signaling pathways and is reportedly cytotoxic to multidrug-resistant tumors (Jia et al. 2004; Popovich and Kitts 2004; Li et al. 2006; Du et al. 2011; Musende et al. 2012; Zhu et al. 2011; Zou et al. 2008). PPD is currently in clinical trials as an anticancer drug candidate (Saklani and Kutty 2008; PanaGin Pharmaceuticals Inc. 2009). PPD is present in trace or undetectable amounts in ginseng plants because of the rapid flux of intermediates toward the final products of ginsenosides. The artificial production of PPD from naturally cultivated ginseng roots requires challenging deglycosylation via enzymatic and chemo-physical treatment (Leung and Wong 2010).
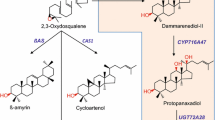
P. ginseng encodes a unique dammarenediol synthase (PgDDS), one of 2,3-oxidosqualene cyclases, catalyzes the production of dammarenediol-II (DD) from 2,3-oxidosqualene, as shown in Fig. 1 (Han et al. 2006; Tansakul et al. 2006). Subsequently, DD is converted to PPD by hydroxylation of dammarenediol-II at C-12 catalyzed by a cytochrome P450 (CYP716A47) (Han et al. 2011). The production of PPD from plant cells or microbial hosts via genetic engineering may be a promising technology for cost-efficient production. Dai et al. (2013) attempted to produce PPD in an engineered yeast expression system. Metabolic engineering of plants is another useful technology for the production of secondary compounds (Verpoorte and Memelink 2002; Wu and Chappell 2008; Dudareva et al. 2013). In this work, we constructed transgenic tobacco co-overexpressing PgDDS and CYP716A47 and determined that the cell suspension culture of transgenic tobacco greatly enhanced PPD production compared with intact tobacco plants.

Biosynthetic pathway for ginsenoside production in P. ginseng. Squalene epoxidase converts squalene to 2,3-oxidosqualene, which is then converted to a triterpene (dammarenediol-II) by dammarenediol synthase (PgDDS). This triterpene subsequently undergoes oxidation by CYP716A47 and CYP716A53v2 and glycosylation and is finally converted to triterpene saponin (ginsenoside)
Results
PPD production in transgenic tobacco co-overexpressing PgDDS and CYP716A47
Transgenic tobacco plants co-overexpressing PgDDS (GenBank accession number, AB122080) and CYP716A47 (GenBank accession number, JN604537) under the control of the CaMV35 promoter were constructed (Fig. 2a). Of the 31 independent transgenic lines, six independent lines were finally selected for analysis (Fig. 2b). Transcription of the BAR, PgDDS, and CYP716A47 genes in the leaves of the transgenic tobacco was confirmed by RT-PCR of cDNA (Fig. 2b). However, no signals were detected in those of wild-type tobacco. The PPD content in the leaf extracts of six transgenic lines was analyzed by gas chromatography-mass spectrometry (GC/MS). The PPD content varied among the transgenic lines, as shown in Fig. 2c. Leaves of transgenic line 6 (Tr6) exhibited the highest PPD content (5.6 µg g−1 DW).

Detection of introduced genes and PPD production in transgenic tobacco co-overexpressing PgDDS and CYP716A47. a Schematic diagram of the PgDDS cDNA under the control of the CaMV35S promoter, which was inserted into pPZP destination vector. pinII terminator: terminator region from Solanum tuberosum proteinase inhibitor II gene, P35S: CaMV 35S promoter, 3′NOS: nopaline synthase terminator. RB represented the right border and LB the left border of the T-DNA. b Expression of the PgDDS, CYP716A47, and bar genes in leaves of wild-type (WT) plants and the indicated transgenic lines (Tr3, Tr5, Tr6, Tr17, Tr20, and Tr31). β-actin was used as a loading control. The wild-type line was not transformed with Agrobacterium. c Content of PPD in transgenic lines. Data are shown as mean values with the standard error bar obtained from three independent plants
Although CYP716A47 catalyzes the conversion of DD to PPD, both DD and PPD accumulated in the tobacco plants. The proportion of PPD to DD varied among the organs, and the accumulation of DD and PPD in the Tr6 transgenic line occurred in an organ-specific manner (roots > leaves > stems > petioles) (Fig. 3a). The content of DD and PPD in the roots was 14.6 and 16.8 µg g−1 DW, respectively (Fig. 3). The GC chromatogram revealed two new products (DD and PPD) at retention times of 39.2 and 44.5 min in all plant organs (Fig. 3b). The two peaks were identical to the retention time of authentic DD and PPD (Fig. 3b). Furthermore, the MS spectra revealed that the fragmentation patterns of PPD (m/z 424) completely matched the MS spectra of authentic PPD (Fig. 3c).

GC-MS analyses of extracts from the various parts of transgenic tobacco (Tr6) co-overexpressing PgDDS and CYP716A47. a Contents of DD and PPD in the leaf, petiole, stem and roots of transgenic tobacco (line 6) co-overexpressing PgDDS and CYP716A47. The data are mean values with the standard deviation from three independent plants. b GC chromatogram of root extracts of transgenic (Tr6) tobacco. Colored lines represent tobacco organs (red petiole; green leaf; back stem; blue root). c MS spectrum of the identified PPD peak obtained from a transgenic line (Tr6). The inset represents the MS spectrum of standard PPD
2,4-D treatment enhances PPD accumulation in the callus
To establish DD and PPD production via cell suspension culture, leaf segments of the transgenic line (TR6) were cultured on solid medium containing various types of growth regulators to induce callus formation. After 2 weeks of culture, the DD and PPD content of the calluses was analyzed by GC/MS. The GC analysis revealed that DD and PPD accumulation was highly stimulated by auxin treatment (Fig. 4). Among the growth regulators, 2,4-D enhanced DD and PPD accumulation was most effectively. Cytokinin treatment did not stimulate DD and PPD accumulation. Combined treatment with auxin and cytokinin was less effective than auxin treatment alone. The increased accumulation of DD and PPD by 2,4-D treatment was clearly correlated with the suppression of phytosterol (campesterol, stigmasterol, and beta-sitosterol) accumulation (Fig. 4). The amount of campesterol, stigmasterol, and beta-sitosterol was the highest in the callus induced by 6-benzyladenine (BA) and lowest in the callus induced by 2,4-D treatment (Fig. 4).

Contents of DD and PPD in the leaf-derived callus of the transgenic tobacco (line 6) co-overexpressing PgDDS and CYP716A47 induced on medium with various growth regulators. Data are shown as mean values with the standard error bar obtained from three independent plants
Stimulation of mevalonate pathway genes by 2,4-D treatment in transgenic tobacco
HMG-Co reductase (HMGR), squalene synthase, and squalene epoxidase are key regulatory enzymes for phytosterol and saponin biosynthesis (Ryder 1991; Chappell et al. 1995; Lee et al. 2004). Cycloartenol synthase, which belongs to a family of 2,3-oxidosqualene cyclases, produces the first carbocyclic intermediate in the sterol pathway in plants (Corey et al. 1993). To address the molecular mechanism by which 2,4-D treatment enhances sapogenin accumulation and reduces phytosterol accumulation, the transcription activities of the tobacco HMGR1 (accession number: U60452), squalene synthase (accession number: U60057), squalene epoxidase (accession number: FS437075), and cycloartenol synthase (accession number: FS401086) were monitored by qPCR (Fig. 5). 2,4-D and IBA treatment enhanced the transcription of HMGR1, squalene epoxidase, and cycloartenol synthase but not squalene synthase. The effects of BA treatment were similar to those of no growth regulator treatment. The transcription of PgDDS and CYP716A47 driven by the 35S promoter was not altered by growth regulators (data not shown).

qPCR of genes upstream of triterpene biosynthesis in the co-overexpressing transgenic roots of P. ginseng. Transcription of HMGR1 (a), tobacco squalene synthase (b), tobacco squalene epoxidase (c), and tobacco cycloartenol synthase (d) in transgenic tobacco (Tr6)
Production of PPD by cell suspension culture
The production of PPD by cell suspension culture of transgenic tobacco is advantageous because it is free from environmental risks associated with the field cultivation of genetically engineered plants. To establish the cell suspension culture, calluses induced from leaf segments of the Tr6 transgenic line were transferred into liquid medium in a 250-ml Erlenmeyer flask and maintained for 3-week subculture intervals. When the suspended cells were reached to friable and homogenous cells clumps after five consecutive sub-cultures as seen in Fig. 6b, cell growth and PPD production were measured. A 12.9-fold increase in the fresh weight was obtained after 3 weeks of culture (Table 1). DD and PPD production reached 32.6 and 689.9 µg g−1 dry weight, respectively (Table 1, Fig. 6). These engineered cells were cultured in 2 l of medium in a 5-l balloon-type bioreactor. A 13.5-fold increase in fresh weight was obtained after 3 weeks of culture (Table 1). DD and PPD production reached 166.9 and 980.9 µg g−1 dry weight, respectively (Table 1, Fig. 6).

GC-MS chromatograms of DD and PPD in shake flask and bioreactor culture of transgenic tobacco cells co-overexpressing PgDDS and CYP716A47. a GC chromatogram extracted from suspended cells of the shake flask culture of non-transgenic tobacco cells. b GC chromatogram of DD and PPD from suspended cells of the shake flask culture in transgenic tobacco (Tr6). c GC chromatogram of DD and PPD from suspended cells of the bioreactor culture in transgenic tobacco (Tr6)
Discussion
PPD production in transgenic tobacco
PPD is a dammarene-type triterpene aglycone ginsenoside and is present in very low levels in natural ginseng roots. This compound is one of the most bioactive compound among many types of ginsenosides and their aglycones (Jia et al. 2004; Popovich and Kitts 2004; Qi et al. 2010). Therefore, metabolic engineering of plants to enable the heterologous expression of triterpene and sapogenin synthase genes is the best strategy for cost-effective production. There are many attempts to increase the ginsenoside production using biotechnological methods in P. ginseng (Murthy et al. 2014). However, most of these works were focused on the production of total ginsenoside saponins which are the products at last step of saponin biosynthesis. The sapogenin aglycones such as PPD are intermediate products and existed in very low amounts in natural ginseng roots. Thus, metabolic engineering is the best protocols for the production of such kinds of sapogenin aglycones.
Sapogenins of ginsenosides are synthesized from DD, which is produced by the oxidosqualene cyclase dammarenediol synthase (PgDDS) (Han et al. 2006). DD is further converted to PPD by a CYP enzyme (CYP716A47) that catalyzes the hydroxylation of DD at the C-12 position (Han et al. 2011). DD and PPD are present in undetectable amounts in the natural roots of ginseng because of the rapid flux of intermediates toward the final production of ginsenosides by glycosyltransferases.
In transgenic tobacco co-overexpressing PgDDS and CYP716A47, DD and PPD were produced at considerable amounts in all tobacco tissues. The ratio of DD and PPD varied among tobacco plant organs. PPD production was greater in the roots than other organs. This result indicates that the root is the best organ for PPD production in intact tobacco plants and that an alternative mechanism may exist for the conversion of DD into PPD. In transgenic plants, the biosynthesis of terpenoids is strictly controlled because terpenoid accumulation occurs in a tissue- and organ-specific manner (Tholl 2006).
Most eukaryotic P450 s are not self-sufficient enzymes, and their catalytic activities strictly rely on an electron donor, NADPH:cytochrome P450 reductase (CPR; Lu et al. 1969). Higher plants have multiple forms of CPR (Durst and Nelson, 1995). Plants appear to use distinct CPR isoforms to meet the high reductive demand of P450-mediated reactions under stressed conditions (Ro et al. 2002). The CYP716A47 gene is strongly responsive to elicitor treatment (methyl jasmonate) (Han et al. 2011). Additional appropriate CPR expression in organs and cells may optimize the conversion of DD to PPD in transgenic tobacco.
Enhanced production of PPD in callus and cell suspension culture
When the leaf segments were cultured on medium containing auxin to induce callus formation, DD and PPD production increased remarkably compared with intact organs of tobacco plants. Treatment with 2,4-D produced much stronger effects than IBA; however, cytokinin (BA) treatment was not effective. GC analysis revealed that the enhanced production of DD and PPD in the leaf-derived callus resulted in reduced phytosterol (β-sitosterol, campesterol, and stigmasterol) content. The reduced phytosterol accumulation in transgenic tobacco may be due to competition for precursors because both phytosterols and triterpenes in plants are synthesized from the common precursor 2,3-oxidosqualene. Alternatively, 2,4-D treatment may regulate upstream genes of triterpene biosynthesis and/or suppress the biosynthesis of phytosterols. The transcription activities of upstream genes (HMGR, squalene synthase, and squalene epoxidase) and cycloartenol synthase genes in tobacco were analyzed by qPCR. Treatment with 2,4-D enhanced the transcription of HMGR, squalene epoxidase, and cycloartenol synthase. HMGR, squalene synthase, and squalene epoxidase are key regulatory enzymes for phytosterol and saponin biosynthesis (Ryder 1991; Chappell et al. 1995; Lee et al. 2004). The enhanced expression of HMGR and squalene epoxidase may have resulted in enhanced production of DD and PPD. However, it is unclear why phytosterol accumulation was strongly reduced even though cycloartenol synthase gene expression was enhanced by 2,4-D treatment. The biosynthesis of phytosterol from cycloartenol involves a number of additional steps. We cannot rule out the suppression of other genes involved in the sterol biosynthetic pathway.
In transgenic tobacco (Tr6), the root extracts contained 14.6 µg g−1 DW of DD and 16.8 µg g−1 DW of PPD. The callus induced from the leaf of the Tr6 line exhibited a 53.7-fold increase in DD production (783.8 µg g−1 DW) and a 7.5-fold increase in PPD production (125.9 µg g−1 DW). When we analyzed the DD and PPD content in the bioreactor culture, the ratio of DD to PPD was highly altered compared with intact tobacco and calluses. The content of DD and PPD was 166.9 and 980.9 µg g−1 DW, respectively, indicating that the suspended cell culture system was highly efficient for the production of PPD compared to intact tobacco plants and calluses. Thus, PPD production via cell suspension culture was the most efficient system for PPD production and may be applied to the large-scale production of these important pharmacologically active compounds.
Materials and methods
Construction of transgenic tobacco co-overexpressing PgDDS and CYP716A47
The open reading frame (ORF) sequence of PgDDS (GenBank accession number, AB122080) was isolated by PCR using the primers 5′-ATG TGG AAG CTG AAG GTT GCT CAA GGA-3′ and 5′-TTA AAT TTT GAG CTG CTG GTG CTT AGG C-3′ (Han et al. 2006). The ORF of CYP716A47 (GenBank accession number, JN604536) was amplified using the PCR primers 5′-ATG GTG TTG TTT TTC TCC CTA TCT-3′ and 5′-TTA ATT GTG GGG ATG TAG ATG AAT-3′. Both genes were cloned via the GATEWAY vector pCR8/GW/TOPO (Invitrogen, Carlsbad, CA) and transferred into the destination vector pPZIP-Bar under the control of the double 35S CaMV promoter (Fig. 2a). Eventually, the overexpression construct harboring both PgDDS and CYP716A47 was introduced into Agrobacterium tumefaciens GV3101 competent cells by the heat shock method.
The construction of transgenic tobacco with the PgDDS and CYP716A47 genes by A. tumefaciens-mediated transformation was essentially the same as described by Han et al. (2014), with the exception of the change of selection medium.
RT-PCR analysis
Total RNA was isolated from tobacco plants using an RNeasy plant mini kit (Qiagen) according to the manufacturer’s instructions and was reverse transcribed using the ImProm-II Reverse Transcription System (Promega, Madison, WI, USA). The first-strand DNA was used as the template for RT-PCR analysis, which was performed as follows: 96 °C for 5 min; 30 cycles of 96 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, and a final 10 min extension at 72 °C. The primers used to amplify the PgDDS gene were 5′-ATG TGG AAG CTG AAG GTT GCT CAA GGA-3′ and 5′-TTA AAT TTT GAG CTG CTG GTG CTT AGG C-3′. The primers used to amplify the CYP716A47 gene were 5′-ATG GTG TTG TTT TTC TCC CTA TCT-3′ and 5′-TTA ATT GTG GGG ATG TAG ATG AAT-3′, and the primers used for the Bar gene were 5′-AGG ACA GAG CCA CAA ACA CC-3′ and 5′-ATG CTT GTA TCC AGC TGC G-3′. The PCR mixture was incubated in a DNA thermal cycler (Applied Biosystems, CA, USA) under the following conditions: one cycle of 94 °C for 5 min, 30 cycles of 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and a final 10-min extension. RT-PCR analysis of β-actin (5′- CGT GAT CTT ACA GAT AGC TTC ATG A-3′ and 5′-AGA GAA GCT AAG ATT GAT CCT CC-3′) was used as the control to confirm RNA integrity and loading accuracy. Electrophoresis of the products was performed using 1 % agar/0.5 % TBA buffer. The RT-PCR analyses were repeated twice, and representative data are presented in the Figs.
Callus induction and real-time PCR (qPCR) analysis
The RNA was isolated from tobacco leaf segments producing callus on medium containing various types of growth regulators (1.0 mg/l IBA, 1.0 mg/l 2,4-D, 1.0 mg/l BA, 1.0 mg/l IBA + 1.0 mg/l BA, 1.0 mg/l 2,4-D + 1.0 mg/l BA). mRNA was reverse transcribed using the ImProm-II Reverse Transcription System (Promega, Madison, WI, USA). Real-time PCR was performed using a Qiagen Rotor Gene Q Real-time PCR detector system with a SYBR Green PCR Kit (Qiagen, Germany). The two-step amplification conditions for all real-time PCR were 95 °C for 5 min, 40 cycles of 95 °C for 5 s, and 60 °C for 10 s. The real-time PCR data shown are the average relative quantities ± SE from at least three replicates. To determine the effect of growth regulators, the expression of each gene was used as the calibrator. The relative expression value of each gene was calculated using the \(^{{ - \varDelta \varDelta C_{\text{T}} }}\) method (Livak and Schmittgen 2001). The tobacco β-actin gene was used for normalization. All primers used in the present study are listed in additional file 1.
Cell suspension culture of transgenic tobacco cells
Leaves of transgenic tobacco were cut into segments 10 mm in length and transferred onto MS medium (Murashige and Skoog 1962) containing various growth regulators (1.0 mg/l 2,4-D, 1.0 mg/l 2,4-D + 1.0 mg/l BA, 1.0 mg/l IBA, 1.0 mg/l IBA + 1.0 mg/l BA, 1.0 mg/l BA), 3 % sucrose, and 0.27 % gelrite that had been adjusted to pH 5.8 and autoclaved at 120 °C for 15 min. Approximately 15 root segments were cultured in a Petri plate for 5 weeks. The culture room was maintained at 22 ± 1 °C under 24 μmol m–2s–1 white florescent lamp illumination.
For the cell suspension culture, a callus derived from the leaf segments was transferred into MS liquid medium containing 1.0 mg/L 2,4-D, 3 % sucrose, and 10 mg/l Basta in a 250-ml Erlenmeyer flask and subcultured for a 3-week interval. To estimate the growth of the cell mass during the culture periods, 8 g of fresh cell mass (packed cell) was transferred to 400 ml of MS liquid medium in a 1000-ml Erlenmeyer flask. For cell suspension culture in balloon-type bubble bioreactors, 20 g of initial cells were transferred to bioreactors (5-l capacity) containing 2 l of MS medium (working volume). Cell growth (fresh weight and dry weight) and DD and PPD production were assessed after 3 weeks of culture.
GC–MS analysis of DD, PPD, and phytosterols
The hexane extract was evaporated and dissolved in methanol (1 ml). A 1-µl aliquot of solution was analyzed by GC (Agilent 7890A) linked to an inert MSD system (Agilent 5975C) with a Triple-Axis detector and equipped with a HP-5MS capillary column (30 m × 0.25 mm, 0.25 mm film thickness). The injection temperature was 250 °C, and the column temperature program was as follows: 150 °C for 5 min, followed by an increase to 300 °C at a rate of 5 °C min−1 and a hold at 300 °C for 20 min. The carrier gas was He, and the flow rate was 1.2 ml min−1. The interface temperature was 300 °C with split injection (10:1). The temperature of the ionization chamber was 250 °C, and ionization mode was electron impact at 70 eV. PPD and phytosterol chemicals used as the standard for GC/MS were purchased from Sigma-Aldrich (St. Louis, MO). The DD was provided by Professor Yong Soo Kwon of the Kangwon National University in Korea.
Author contribution statement
Y.E.C designed the experiments. P.B.A constructed the transgenic tobacco. S.B.P and J.H.C. performed RT-PCR and qPCR. J.Y.H performed the GC/MS analysis. Y.E.C prepared the figures and wrote the article.
Abbreviations
- BAR:
-
Basta
- DD:
-
Dammarenediol-II
- PPD:
-
Protopanaxadiol
- DDS:
-
Dammarenediol-II synthase
- PgDDS:
-
Panax ginseng dammarenediol-II synthase
- CYP716A47:
-
Cytochrome P450 716A47
- qPCR:
-
Real-time polymerase chain reaction
References
Bae EA, Han MJ, Kim EJ, Kim DH (2004) Transformation of ginseng saponins to ginsenoside Rh2 by acids and human intestinal bacteria and biological activities of their transformants. Arch Pharm Res 27:61–67
Chappell J, Wolf F, Proulx J, Cuellar R, Saunders C (1995) Is the reaction catalyzed by 3-hydroxy-3-methylglutaryl coenzyme A reductase a rate-limiting step for isoprenoid biosynthesis in plants? Plant Physiol 109:1337–1343
Corey EJ, Matsuda SP, Bartel B (1993) Isolation of an Arabidopsis thaliana gene encoding cycloartenol synthase by functional expression in a yeast mutant lacking lanosterol synthase by the use of a chromatographic screen. Proc Natl Acad Sci USA 90:11628–11632
Dai Z, Liu Y, Zhang X, Shi M, Wang B, Wang D, Huang L, Zhang X (2013) Metabolic engineering of Saccharomyces cerevisiae for production of ginsenosides. Metab Eng 20:146–156
Du GJ, Dai Q, Williams S, Wang CZ, Yuan CS (2011) Synthesis of protopanaxadiol derivatives and evaluation of their anticancer activities. Anticancer Drugs 22:35–45
Dudareva N, Klempien A, Muhlemann JK, Kaplan I (2013) Biosynthesis, function and metabolic engineering of plant volatile organic compounds. New Phytol 198:16–32
Durst F, Nelson DR (1995) Diversity and evolution of plant P450 and P450-reductases. Drug Metabol Drug Interact 12:189–206
Han JY, Kwon YS, Yang DC, Jung YR, Choi YE (2006) Expression and RNA interference-induced silencing of the dammarenediol synthase gene in Panax ginseng. Plant Cell Physiol 47:1653–1662
Han JY, Kim HJ, Kwon YS, Choi YE (2011) The cyt P450 enzyme CYP716A47 catalyzes the formation of protopanaxadiol from dammarenediol-II during ginsenoside biosynthesis in Panax ginseng. Plant Cell Physiol 52:2062–2073
Han JY, Wang HY, Choi YE (2014) Production of dammarenediol-II triterpene in a cell suspension culture of transgenic tobacco. Plant Cell Rep 33:225–233
Hasegawa H, Sung JH, Benno Y (1997) Role of human intestinal Prevotella oris in hydrolyzing ginseng saponins. Planta Med 63:436–440
Jia W, Yan H, Bu X, Liu G, Zhao Y (2004) Aglycone protopanaxadiol, a ginseng saponin, inhibits P-glycoprotein and sensitizes chemotherapy drugs on multidrug resistant cancer cells. ASCO annual meeting proceedings (post-meeting edition). J Clin Oncol 22(Suppl):9663
Karikura M, Miyase T, Tanizawa H, Taniyama T, Takino Y (1991) Studies on absorption, distribution, excretion and metabolism of ginseng saponins. VII. Comparison of the decomposition modes of ginsenoside-Rb1 and -Rb2 in the digestive tract of rats. Chem Pharm Bull 39:2357–2361
Lee MH, Jeong JH, Seo JW, Shin CG, Kim YS, In JG, Yang DC, Yi JS, Choi YE (2004) Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol 45:976–984
Leung KW, Wong AS (2010) Pharmacology of ginsenosides: a literature review. Chin Med 5:20
Li G, Wang Z, Sun Y, Liu K, Wang Z (2006) Ginsenoside 20(S)-protopanaxadiol inhibits the proliferation and invasion of human fibrosarcoma HT1080 Cells. Basic Clin Pharmacol Toxicol 98:588–592
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using realtime quantitative PCR and the \(2^{{ - \varDelta \varDelta C_{\text{T}} }}\) Method. Methods 25:402-408
Lu AY, Junk KW, Coon MJ (1969) Resolution of the cytochrome P450 containing omega-hydroxylation system of liver microsomes into three components. J Biol Chem 244:3714–3721
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio-assays with tobacco tissue cultures. Physiol Plant 15:473–497
Murthy HN, Georgiev MI, Kim YS, Jeong CS, Kim SJ, Park SY, Paek KY (2014) Ginsenoside: prospective for sustainable biotechnolotgical production. Appl Microbiol Biotechnol 98:6243–6254
Musende AG, Eberding A, Wood CA, Adomat H, Fazli L, Hurtado-Coll A, Jia W, Bally MB, Tomlinson Guns ES (2012) A novel oral dosage formulation of the ginsenoside aglycone protopanaxadiol exhibits therapeutic activity against a hormone-insensitive model of prostate cancer. Anticancer Drugs 23:543–552
PanaGin Pharmaceuticals Inc (2009) http://www.panagin.com. Accessed Oct 2009
Popovich DG, Kitts DD (2004) Ginsenosides 20(S)-protopanaxadiol and Rh2 reduce cell proliferation and increase sub-G1 cells in two cultured intestinal cell lines (Int-407 and Caco-2). Can J Physiol Pharmacol 82:183–190
Qi LW, Wang CZ, Yuan CS (2010) American ginseng: potential structure–function relationship in cancer chemoprevention. Biochem Pharmacol 80:947–954
Ro D, Ehlting J, Douglas C (2002) Cloning, functional expression, and subcellular localization of multiple NADPH-cytochrome P450 reductases from hybrid poplar. Plant Physiol 130:1837–1851
Ryder NS (1991) Squalene epoxidase as a target for the allylamines. Biochem Soc Trans 19:774–777
Saklani A, Kutty SK (2008) Plant-derived compounds in clinical trials. Drug Disc Today 13:161–171
Shibata S (2001) Chemistry and cancer preventing activities of ginseng saponins and some related triterpenoid compounds. J Korean Med Sci 16:S28–S37
Tansakul P, Shibuya M, Kushiro T, Ebizuka Y (2006) Dammarenediol-II synthase, the first dedicated enzyme for ginsenoside biosynthesis, in Panax ginseng. FEBS Lett 580:5143–5149
Tawab MA, Bahr U, Karas M, Wurglics M, Schubert-Zsilavecz M (2003) Degradation of ginsenosides in humans after oral administration. Drug Metab Dispos 31:1065–1071
Tholl D (2006) Terpene synthases and the regulation, diversity and biological roles of terpene metabolism. Curr Opin Plant Biol 9:1–8
Verpoorte R, Memelink J (2002) Engineering secondary metabolite production in plants. Curr Opin Biotech 13:181–187
Wu S, Chappell J (2008) Metabolic engineering of natural products in plants; tools of the trade and challenges for the future. Curr Opin Biotech 19:145–152
Zhu GY, Li YW, Tse AK, Hau DK, Leung CH, Yu ZL, Fong WF (2011) 20(S)-protopanaxadiol, a metabolite of ginsenosides, induced cell apoptosis through endoplasmic reticulum stress in human hepatocarcinoma HepG2 cells. Eur J Pharmacol 668:88–98
Zou W, Yue P, Khuri FR, Sun SY (2008) Coupling of endoplasmic reticulum stress to CDDO-Me-induced up-regulation of death receptor 5 via a CHOP-dependent mechanism involving JNK activation. Cancer Res 68:7484–7492
Acknowledgments
This work was supported by the Rural Development Administration, Republic of Korea [Next-Generation BioGreen 21 Program (PJ011285)], and by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2013R1A1A2064352).
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Sato.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chun, JH., Adhikari, P.B., Park, SB. et al. Production of the dammarene sapogenin (protopanaxadiol) in transgenic tobacco plants and cultured cells by heterologous expression of PgDDS and CYP716A47 . Plant Cell Rep 34, 1551–1560 (2015). https://doi.org/10.1007/s00299-015-1806-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-015-1806-9