
Abstract
This review provides an overview of SAPHO (Synovitis, Acne, Pustulosis, Hyperostosis, and Osteitis), a rare autoinflammatory disease that primarily affects bones, skin, and joints. We conducted a search on Medline/PubMed using keywords such as SAPHO syndrome, chronic recurrent multifocal osteitis/osteomyelitis, and related terms. SAPHO syndrome is rare, with a reported frequency of 1 in 10,000 in the Caucasian population. However, the actual incidence of SAPHO syndrome is unknown, and the incidence of the disease is likely higher. The pathogenesis of SAPHO syndrome remains incompletely understood. Current evidence suggests that SAPHO results from a complex interplay between immune dysregulation, genetic susceptibility, and environmental factors. It's not clear if SAPHO syndrome is an autoimmune disease or an autoinflammatory disease, but current evidence suggests that it's more likely an autoinflammatory disease because of things like neutrophil hyperactivity, fewer natural killer (NK) cells, high levels of interleukin (IL)-1, and a good response to treatments that block IL-1. Osteo-articular (OA) involvement is a key clinical feature of SAPHO. It affects the anterior chest wall, axial skeleton, peripheral joints, mandible, long bones of the extremities, and pelvis. Dermatological involvement is a common target in SAPHO, with lesions observed in 60–90% of cases. Common skin lesions include psoriasis and acne, with hidradenitis suppurativa and neutrophilic dermatoses being less commonly seen. Other clinical findings include constitutional symptoms caused by systemic inflammation, such as fever, weight loss, and fatigue. There is no specific laboratory finding for SAPHO syndrome. However, during active disease, there may be an increase in positive acute phase markers, such as erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), complement levels, mild leukocytosis, and thrombocytosis. Diagnosis is crucial for SAPHO syndrome, which lacks a specific diagnostic finding and is often underrecognized. A comprehensive evaluation of a patient's medical history and physical examination is crucial. Treatment options include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, conventional and synthetic disease-modifying agents (cDMARDs and sDMARDs), biological therapies, bisphosphonates, and antibiotics. Biological treatments have emerged as a viable alternative for SAPHO patients who do not respond to conventional treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
SAPHO syndrome, an acronym for synovitis, acne, pustular lesions, hyperostosis, and osteitis, is a rare, complex, and chronic inflammatory rheumatic disease that often follows a relapsing course [1,2,3,4]. First identified in the late 1980s, SAPHO syndrome has been associated with various manifestations, including acne-associated arthritis and sterno-clavicular hyperostosis (SCH) [5]. The syndrome shares clinical and radiological features with certain childhood diseases, such as chronic recurrent multifocal osteomyelitis (CRMO), recurrent osteomyelitis of the jaw, and Köhler's disease, which are considered its variants [3, 6, 7]. Despite being a rare condition, SAPHO syndrome has been receiving increasing attention in clinical and research settings due to the significant challenges it presents for clinicians in terms of diagnosis and treatment. The syndrome's diverse clinical presentations and tendency for relapse make it particularly difficult to manage. The purpose of this review is to offer a comprehensive and detailed understanding of SAPHO syndrome, encompassing its epidemiology, etiology, clinical manifestations, and management strategies (Fig. 1). By increasing awareness and knowledge, this review aims to improve patient outcomes for those affected by this condition.

Summarizing innovative points of the review
Search strategy and methods
We conducted a search on Medline/PubMed, Scopus, and Embase using keywords such as SAPHO syndrome, chronic recurrent multifocal osteitis/osteomyelitis, and related terms. Publications between 1983 and 2023 were scanned. We included case reports, case–control studies, and retrospective cohort studies in our search strategy. Then, non-English publications, duplicates, and publications that did not comply with the aim of our review were excluded from the review. We also extracted additional relevant articles from the references of the retrieved papers [8]. It covers the epidemiology, clinical presentation, pathogenesis, and various treatment options available.
Epidemiology
SAPHO syndrome is a rare, complex, and chronic inflammatory rheumatic disease with a reported frequency of 1 in 10,000 in the Caucasian population [1, 4]. However, the actual incidence of SAPHO syndrome is unknown, and the incidence of the disease is likely higher, as it is often misdiagnosed or classified under a different illness, leading to underreporting [9]. SAPHO syndrome primarily affects individuals aged 30 – 50 and is more commonly observed in females [10,11,12,13]. However, cases have been reported in children and elderly individuals over 80 years old [10,11,12,13].
Pathogenesis
The pathogenesis of SAPHO syndrome remains incompletely understood, despite considerable research efforts. Current evidence suggests that SAPHO results from a complex interplay between immune dysregulation, genetic susceptibility, and environmental factors [3, 6,14]. While the presence of the disease in certain family members in some studies suggests a genetic predisposition, no specific gene has definitively been linked to this disease [15, 16]. Some studies have proposed a potential association with human leukocyte antigens (HLA), such as HLA-A26, HLA-B27, HLA-B39, and HLA-B61, but the relationship between these genes and SAPHO, particularly HLA-B27, has not been confirmed [1, 3, 14, 17]. Additionally, some nucleotide polymorphisms (MDM2 T309G, p53 G72C, rs6908425, and CDKAL1 T > C) have been suggested to be associated with SAPHO, but compelling evidence to support this hypothesis remains lacking [3, 18, 19]. While diseases involving certain genes on chromosomes 1 and 18 (LPIN2, PSTPIP2, and NOD2) have been suggested to share clinical features with SAPHO syndrome, these associations have not been observed in individuals with the condition [3, 19].
In the development of SAPHO syndrome, infections are considered to be one of the most significant environmental factors. Tissue samples taken from bone and joint lesions of SAPHO patients have revealed the presence of various microorganisms, including Cutibacterium acnes (C. acnes), formerly Propionibacterium, Staphylococcus aureus, Haemophilus parainfluenzae, Treponema pallidum, and Actinomycetes, suggesting their potential role in the development of the disease [14] Among these microorganisms, C. acnes, a Gram-positive, anaerobic, slow-growing bacterium, is considered the most crucial. C. acnes primarily resides commensally in sebaceous glands and, to a lesser extent, in the intestines [14]. According to a study, 42% of bone biopsies from individuals with SAPHO syndrome were found to contain this microorganism. However, detecting the presence of C. acnes in SAPHO patients can be challenging due to the microorganism's slow growth, and the need for advanced laboratory techniques such as PCR and expertise in its detection, which could potentially increase this rate [20, 21]. Certain studies have investigated how C. acnes can lead to SAPHO syndrome [14, 22, 23]. These studies propose that due to a genetic or molecular defect that impairs its elimination, the microorganism can grow uncontrollably and subsequently produce various enzymes and metabolites that can directly or indirectly damage host cells. It has been hypothesized that this situation may be linked to lower levels of natural killer (NK) cells responsible for eliminating C. acnes in SAPHO patients compared to healthy controls, the suppression of the Forkhead Box O1 (FoxO1) protein responsible for clearing the bacterium through various mechanisms such as Western-style diets and androgens, and the activation of complement by C. acnes, leading to the release of proinflammatory cytokines such as interleukin (IL)-1, IL-8, IL17-A and tumor necrosis factor (TNF)-α [22,23,24]. These cytokines can affect both cellular and humoral proinflammatory pathways, as depicted in Fig. 2 [14, 23, 24]. Furthermore, an experimental study demonstrated that injecting C. acnes into the joint regions of rats led to erosion in the joint area as well as proliferation of synovial cells, highlighting the significant impact of this microorganism on joint health [25].

Keys to unlock pathogenesis; known pathways in the pathogenesis of SAPHO syndrome
The classification of SAPHO syndrome as either an autoimmune or autoinflammatory disease remains uncertain. However, current evidence suggests that SAPHO is more likely an autoinflammatory condition due to various factors, including neutrophil hyperactivity, reduced natural killer (NK) cells, elevated levels of interleukin (IL)-1, and a positive response to anti-IL-1 treatments [26]. Furthermore, SAPHO syndrome shares similarities with several autoinflammatory syndromes, including Majeed, DIRA (deficiency of IL-1 receptor antagonist), PAPA (pyogenic arthritis, pyoderma gangrenosum, acne), and PASS (pyoderma gangrenosum, acne vulgaris, hidradenitis suppurativa, and ankylosing spondylitis). These diseases display various degrees of similarity to SAPHO and respond well to anti-IL-1 treatments (Table 1) [27,28,29,30]. In addition, animal models have supported the autoinflammatory origin of SAPHO syndrome, with mice lacking PSTPIP2 exhibiting clinical similarities to SAPHO, such as synovitis, hyperostosis, osteitis, and multifocal osteomyelitis; macrophage and neutrophil accumulation in bone, joints, and skin; as well as elevated blood levels of IL-1. Notably, these mice also display an overgrowth of the Prevotella bacterium in their intestines, which is associated with increased production of IL-1β, resembling the relationship between SAPHO and C. acnes [14, 31]. Therefore, it is likely that SAPHO syndrome belongs to the autoinflammatory disease group.
Abnormal cytokine levels (such as IL-1, IL-8, IL-17, IL-18, and TNFα) in individuals with SAPHO syndrome suggest the involvement of the Th-17 pathway [22, 26, 32]. A proposed mechanism for the inflammation seen in SAPHO involves an imbalance between Th-17 and regulatory T cells (T-reg) caused by a reduction in NK cells [24]. This leads to the stimulation of the innate immune system due to the inability to eliminate C. acnes, ultimately resulting in chronic inflammation in SAPHO syndrome through various mechanisms, including T-cell-mediated immune processes [22, 30].
Macrolide antibiotics, such as azithromycin, have been demonstrated to be effective in treating SAPHO syndrome, particularly in improving skin manifestations and, to a certain extent, osteoarticular (OA) symptoms [33, 34]. Nonetheless, relapses are common in most cases after discontinuing the medication, indicating that while the infectious process contributes to the disease's development, other mechanisms are also involved in its persistence [33, 34].
Histopathology
Histological evaluations of tissue samples taken from SAPHO patients have revealed that in the early stages of the disease, the bone exhibits periosteal bone reactions and histological findings similar to those observed in osteomyelitis. As the disease progresses, chronic inflammation leads to the accumulation of mononuclear cells, and in later stages, bone marrow fibrosis, sclerosis, and enlarged trabecular structures are observed [32].
Clinical findings
SAPHO syndrome mainly affects the bones, joints, and skin, as suggested by its acronym (synovitis, acne, pustular lesions, hyperostosis, and osteitis). However, not all patients display all of these clinical findings, and the severity and frequency of symptoms may vary. Some individuals may experience mild symptoms, while others may have a more severe disease course. Symptoms can appear in single or multiple areas and may occur continuously or episodically with periods of flares and remission. Ultimately, clinical symptom severity is influenced by location, extent, and disease activity [1, 4, 17].
Osteo-articular (OA) involvement
Bone and joint complaints are the key clinical features of SAPHO disease and are seen in all patients, regardless of the presence or absence of dermatological findings. OA involvement can manifest as osteitis, hyperostosis, synovitis, or a combination of these [2]. Enthesitis, inflammation of tendons at the insertion points of bones, is also a significant clinical finding in SAPHO patients. Osteitis is characterized by tenderness or swelling in the affected areas, with inflammatory changes occurring in the cortex, medulla, or both. Hyperostosis is defined as excessive bone growth that occurs in the later stages of the disease due to endosteal or periosteal reactions.
OA involvement most commonly affects the anterior chest wall, axial skeleton (spine and sacroiliac joints [SIJ]), peripheral joints, mandible, long bones of the extremities, and pelvis based on the rate of frequency [3, 35]. Anterior chest wall involvement is a characteristic finding of SAPHO and affects 65–90% of cases. In this region, sterno-costal, sterno-clavicular, and osteo-clavicular ligaments are affected [4, 9]. Anterior chest wall involvement can be divided into three stages: the first stage is limited enthesopathy in the costo-clavicular ligament; the second stage is the development of osteolytic and osteosclerotic changes in the sterno-clavicular joint and involvement of surrounding structures; and the final stage is the involvement of areas with osteosclerosis and hyperostosis development [36]. Axial involvement occurs in about half of patients and can affect the spine or SIJ, with thoraco-lumbar spinal involvement being the most common [37]. Sacroiliitis occurs in about one-third of cases and causes inflammatory-type pain in the lumbar or gluteal region seen more frequently than lumbar involvement [11]. SIJ involvement usually presents as unilateral osteitis with synovitis developing over time. Joint inflammation is the main finding in synovitis, which can present as arthritis affecting the knee, hip, or ankle joints [11, 12]. An oligo-articular involvement pattern is generally observed, and joint involvement is usually non-erosive, although some cases may exhibit peri-articular osteopenia, erosion, and narrowing of the joint space [16, 38]. Mandibular involvement occurs in approximately 10% of cases, typically affecting the body of the mandible, and presents clinically as recurrent sterile osteomyelitis [35, 39, 40]. Tibia and femur involvement are the most common among long bones, with a frequency of about 5% reported. In addition, pelvic and symphysis pubis involvement (approximately 3%) are also among the OA involvement findings [38, 39, 41].
Dermatological involvement
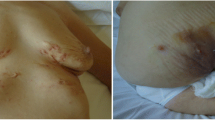
Skin is a common target in SAPHO, and lesions can be observed in 60–90% of cases. Lesions may appear before (50%), simultaneously with (20%), or after (30%) OA findings [3, 17]. Compared to OA findings, skin findings tend to be more severe and resistant to treatment. Exacerbations of skin and OA findings may occur independently of each other [3]. The most common skin lesions are psoriasis and acne, with hidradenitis suppurativa and neutrophilic dermatoses, including Sneddon-Wilkinson (subcorneal pustular dermatosis), pyoderma gangrenosum, and Sweet syndrome, being less commonly seen [4]. One individual may have two or three different types of skin involvements, with palmoplantar pustulosis (PPP) and psoriasis vulgaris being the most common co-occurrences [4]. SAPHO was once considered a subgroup of psoriatic arthritis (PsA) due to its strong association with psoriasis, and approximately 3% of PsA patients have been reported to have SAPHO. PPP is the most common skin lesion and occurs alone or with other skin findings in approximately 90% of cases with skin involvement. Severe acne (15%) and psoriasis vulgaris (14%) are also observed.
Other clinical findings
Constitutional symptoms caused by systemic inflammation, such as fever, weight loss, and fatigue, have been reported in some cases. Inflammatory bowel disease, particularly Crohn's disease, can occur in around 10% of SAPHO patients [42]. Venous thrombosis is another clinical finding, occurring in approximately 2% of patients in one case series [43]. A computed tomography study evaluating pulmonary findings suggested that pulmonary opacities were observed more frequently in SAPHO patients compared to the healthy group [44]. Although rare, scleritis, uveitis, and other eye findings, hypertrophic pachymeningitis, and AA amyloidosis have also been reported [45].
Radiographic findings
SAPHO syndrome is characterized by various bone lesions that can be visualized with radiographic examination. Bone lesions tend to be osteolytic in the early stages and osteosclerotic in the advanced stages. Radiographic lesions that can be visualized include osteolysis, osteitis, osteosclerosis, and hyperostosis. Currently, conventional X-rays, computed tomography (CT), bone scintigraphy, magnetic resonance imaging (MRI), and positron emission tomography (PET) are used for imaging in SAPHO syndrome [46].
Conventional radiography is the most commonly used imaging technique due to its availability and lower cost. However, it has limitations in detecting pathological findings in the early stages and inadequate evaluation of anatomical structures due to low resolution [47].
CT provides better evaluation of anatomical structures, especially anterior chest wall structures such as sternoclavicular, upper costal-sternal, and manubriosternal joints, long bones, and spinal involvement. Detection of bone hypertrophy in the region where the costoclavicular ligament attaches at a relatively early stage offers an important diagnostic clue [16, 37].
MRI is useful for detecting early-stage changes such as synovitis and edema and determining lesion activity. Another advantage of MRI is that it can be used for treatment or lesion follow-up due to the absence of radiation risk [48].
Scintigraphy is a useful diagnostic method in SAPHO syndrome, allowing the determination of lesion distribution and frequency [36]. SAPHO lesions are characterized by increased uptake on scintigraphy. This is similar for both active and chronic lesions, so information about lesion activity cannot be obtained from scintigraphy alone. In a scintigraphy study, the anatomical distribution of SAPHO lesions was reported as follows: anterior chest wall 99.4%, spine 59.2%, peripheral joints 34.4%, sacroiliac joint 29.3% (mostly unilateral), and head region 10% (most commonly in the maxilla and mandible). In particular, symetrical involvement in the sternoclavicular joint region, also called the "bull's head," is considered characteristic of SAPHO and can be observed in approximately 10% of cases [36]. This appearance is particularly helpful in diagnosing patients with atypical clinical courses.
PET imaging can provide information on the evaluation of metastatic lesions and lesion activity. This imaging modality is helpful for evaluating the involved sites of SAPHO syndrome [49].
In summary, each imaging modality has its advantages and limitations, and the choice of imaging technique depends on the clinical presentation, the stage of the disease, and the need for assessing specific structures or lesion activity. Early detection and proper evaluation of affected areas are essential for accurate diagnosis and appropriate management.
Laboratory findings
At present, there is no laboratory finding that is specific to SAPHO syndrome. However, during active disease, there may be an increase in positive acute phase markers. About half of the patients in the case series have reported elevated levels of erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP), while increased complement levels, mild leukocytosis, and thrombocytosis have also been suggested during the course of the disease [12].
Various studies on HLA-B27 have reported a frequency of around 2–13%, which is not significantly higher than that of the general population [17]. Rheumatoid factor (RF) and anti-cyclic citrullinated peptide antibody (anti-CCP) are usually negative. Some patients may have increased levels of serum amyloid A protein, and anti-nuclear antibody have been reported to be around 5–20%. Also, anticardiolipin antibodies were positive in a minority of patients [50]. No specific auto-antibody type has been identified yet [51].
Some studies have suggested that during the active phase of SAPHO syndrome, approximately one-quarter of cases exhibit increased levels of IgG4, as well as elevated levels of various cytokines, such as TNF-α, IL-6, IL-8, IL-17A, and IL23 [22, 52]. Additionally, markers of bone metabolism, such as an increase in the osteoclast marker β-CTX (β-isolated C-terminal peptide) and a decrease in osteoblast-derived osteocalcin, have been reported. Patients during the active phase of the disease have also shown an increase in RANKL (receptor activator of nuclear factor kappa-B ligand) and a decrease in the RANKL/osteoprotegerin ratio [26].
Diagnosis
SAPHO syndrome lacks a specific diagnostic finding and is often under-recognized, leading to delayed diagnosis and treatment. A comprehensive evaluation of a patient's medical history and physical examination is crucial for diagnosis. Various diagnostic criteria have been proposed for SAPHO syndrome, with the criteria by Kahn and colleagues being the most widely used [7]. The presence of symptoms such as osteitis, hyperostosis, and synovitis are essential for diagnosing SAPHO. Anterior chest wall involvement or recurrent sterile osteomyelitis should raise suspicion of SAPHO. Skin findings may not be present in all patients, but the presence of neutrophilic dermatoses (especially PPP), severe nodular acne, and psoriasis, when combined with other clinical findings, can be helpful for diagnosis [1, 3] Additionally, the presence of inflammatory bowel disease (IBD) can provide additional clues for diagnosis [1, 17]. For young patients with an unclear diagnosis, whole-body MRI examinations can be helpful. PET scans can provide additional benefits when there is a clinical suspicion of malignancy [12, 49, 53].
Differential diagnosis
When considering the differential diagnosis for SAPHO syndrome, infections and malignancies should be kept in mind. Osteomyelitis should be considered if the clinical picture includes fever and solitary lesions, unlike the multifocal lesions observed in SAPHO syndrome [54, 55]. Malignancies such as osteosarcoma, Ewing's sarcoma, lymphoma, and tumors with bone metastases should also be considered. Localized bone pain, warmth, swelling, tenderness, and the absence of systemic symptoms are similarities between SAPHO and osteosarcoma. However, osteosarcoma usually occurs in a single bone and is characterized by specific radiographic changes. Bone metastases are common in the vertebrae, pelvis, sacrum, and femur and should also be considered. Therefore, in patients who do not respond to treatment, have additional comorbid diseases (such as endocarditis or IV drug users), or have accompanying systemic findings, these possibilities should be considered. Further radiographic evaluations such as biopsy, culture, and PET may be necessary [56].
Since the anterior chest wall is a common site of involvement in SAPHO, it is essential to consider other conditions that can also affect this area (Fig. 3). Tietze's syndrome, which typically presents in young adults with unilateral pain and swelling in the 2–4 costochondral joints, should be considered. The affected areas have pain and swelling, and respiration can cause pain that radiates to the shoulder. While the sternoclavicular joint is rarely involved in Tietze's syndrome, acute phase reactants can be elevated, especially during the active phase [57].

Common causes of sternoclavicular swellings
Besides SAPHO syndrome, RA and PMR can also affect the sternoclavicular region. In RA patients, approximately 20% of cases show involvement in this area, with about 5% of healthy controls presenting asymptomatic swelling. Involvement in this region has also been reported in one-quarter of PMR patients evaluated with PET [58]. Another disease to consider is condensing osteitis, which presents with sclerosis at the medial end of the clavicle and clinical detection of pain and swelling in this area. Distinguishing features include being a non-inflammatory condition, unilateral involvement, joint preservation, and the absence of involvement in any other anterior chest wall structure. Friedrich's disease, characterized by osteonecrosis at the medial end of the clavicle, is another disease affecting this region. It is generally unilateral, with swelling and mild pain present clinically, and rheumatological inquiries and inflammatory parameters being normal. Septic arthritis can involve the sternoclavicular joint in about 1% of cases [56, 59].
Differential diagnosis in the pediatric age group includes Ewing’s sarcoma, histiocytosis, and Majeed syndrome [54].
SAPHO and spondyloarthritis relationship
In recent years, there has been growing interest in exploring the possible relationship between SAPHO and SpA, given their several clinical, radiographic, and pathogenetic similarities. Both conditions can involve the spine and sacroiliac joint, cause peripheral arthritis and enthesitis, and be associated with extra-articular findings such as psoriasis and IBD [3]. Furthermore, the potential relationship with pathogens is seen in both conditions, as the exacerbation of SAPHO symptoms following the discontinuation of antibiotic treatment suggests a possible link with reactive arthritis [33]. Radiographically, both conditions can exhibit sacroiliitis, syndesmophyte development in the spine, and new bone formations. However, unlike SpA, SAPHO does not show an increased frequency of HLA-B27, which is a genetic marker commonly associated with SpA [17]. Despite these similarities, further research is necessary to better understand the relationship between SAPHO and SpA and clarify their underlying pathophysiology.
SAPHO and CRMO relationship
CRMO (Chronic Recurrent Multifocal Osteomyelitis) is a rare autoinflammatory disease that primarily affects children. It is a subtype of nonbacterial osteomyelitis, and it is characterized by sterile and recurrent osteomyelitis attacks [6]. Although C. acnes is the most commonly detected microorganism, no other microorganisms have been found to cause the disease. It typically affects the 7–12 age group, with a female predominance, and the main clinical symptom is insidious onset bone pain. Swelling and an increase in temperature may also be observed at the site of involvement, which tends to be multifocal. Although any bone can be affected, the metaphyses of the long bones of the lower extremities are the most commonly involved sites, followed by the pelvis, vertebrae, clavicles, long bones of the upper extremities, and mandible [6, 30] The pathogenesis of CRMO includes osteoclast activation, bone destruction, hyperostosis, and sclerosis. Similar to SAPHO, CRMO can also involve the skin (PPP, acne, and psoriasis), bowel, and joints, and there is no increase in HLA-B27 frequency [30]. Due to these similarities, CRMO is considered the childhood type of SAPHO disease. Pathological fractures may occur in the affected areas, especially in the vertebrae, and growth retardation can also occur due to bone involvement. Lesions may be overlooked on direct radiographs, and whole-body MRI examination is the preferred imaging method [60, 61].
Prognosis
SAPHO does not have a cure at present. However, it generally has a favorable course, and long-term remissions are possible. There is currently no reliable marker available to predict the prognosis of the disease [1, 4]. It has been reported that the prognosis may be worse in cases involving pathological fractures in the vertebrae or clavicles [13].
Treatment
The primary goal of treating SAPHO is to improve clinical symptoms, enhance quality of life, and prevent disease progression. However, due to the rarity of the disease, there are no controlled studies available, and there is no established treatment algorithm. The available data is based on case series and experiences from open studies. Current treatment options for SAPHO include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, conventional and synthetic disease-modifying agents (cDMARDs and sDMARDs, respectively), biological therapies, bisphosphonates, and antibiotics [62,63]. The treatment decision is influenced by the primary affected area of the disease (skin or osteoarticular), disease activity, and the presence of any concurrent medical conditions or comorbidities [62, 63].
NSAIDs are commonly the initial treatment choice for patients with SAPHO who have osteoarticular symptoms [1, 3]. However, they are often not sufficient in controlling the disease, particularly in cases with widespread involvement. There is no clear superiority among different NSAIDs, and to determine if a patient is non-responsive to NSAIDs, it is recommended to use the full dose of the drug for at least 1 month and preferably try two different NSAIDs. However, NSAIDs are generally ineffective in cases with widespread involvement [64].
Corticosteroids are commonly used for short-term treatment of various OA findings, such as arthritis, in patients who do not respond to NSAIDs [4]. However, long-term use is not recommended due to potential side effects. Corticosteroids can be given systemically or directly into the joint (intra-articularly, IA). In a case series, a single 20 mg dose of triamcinolone was administered to the sternoclavicular region. This resulted in partial effectiveness in treating clinical osteitis at the 3-month assessment, but there was no significant impact on MRI osteitis scores [65].
Conventional DMARDs, such as methotrexate (MTX), are frequently used to treat SAPHO, particularly in cases with peripheral joint involvement. However, their effectiveness in treating osteomyelitis, osteitis, and enthesitis is not well established [9]. Sulfasalazine (SSZ), hydroxychloroquine, leflunomide, cyclosporine, and colchicine treatments have been used in cases with arthritis, but their effectiveness is currently unclear [62]. There is no established treatment algorithm for axial disease and enthesitis, and based on experiences from axial SpA treatment, conventional DMARDs may be ineffective. Nonetheless, due to reimbursement policies in many countries, it is recommended to use DMARDs like MTX or SSZ after NSAIDs (± systemic or IA corticosteroids) before initiating more advanced treatments, such as biological therapies. MTX has also been reported to have some effectiveness on pleural effusion [1, 3].
A comprehensive literature review indicates that approximately 70% of patients who undergo treatment involving a combination of DMARDs and NSAIDs may experience improvements in SAPHO symptoms [66]. However, due to the remaining proportion of patients not achieving desired outcomes, there is a recognized need to explore additional pharmacological interventions.
Bisphosphonates exert a dual action by inhibiting osteoclast activity and bone resorption while also displaying anti-inflammatory effects through the suppression of cytokines such as IL-1, TNF-α, and IL-6. In the context of SAPHO syndrome, both IV bisphosphonates like pamidronate and zoledronic acid, as well as oral bisphosphonates such as alendronate, have been employed as treatment modalities [67]. Notably, a retrospective study involving 14 DMARD-resistant patients explored the use of IV pamidronate treatment. Administering a dosage of 60 mg/day over 3 consecutive days resulted in a significant reduction of over 50% in VAS scores, indicative of bone pain disappearance. This study primarily focused on patients with skin and anterior chest wall involvement, where nearly 90% of cases exhibited sustained improvement following a single infusion cycle. However, it's important to note that two cases displayed incomplete responses, even after a subsequent second cycle. While the treatment effectively targeted OA lesions, the response of skin lesions remained unsatisfactory [68]. Another uncontrolled study involving 30 patients investigated the impact of administering 1 mg/kg/day IV pamidronate for 3 days, with a repeat cycle after 3 months. This approach yielded an 80% effective response rate after 12 months in both clinical and MR spine lesions. However, detailed information regarding the dermatological effects was not provided [69]. In a retrospective study involving 30 patients, IV zoledronic acid was administered at a dosage of 4 mg/day for 3 days. Notably, half of the patients had a history of prior DMARD or biologic use. The study demonstrated significant reductions in VAS, BASDAI, and BASFI scores at the 12-month mark compared to baseline. Comparisons with previous pamidronate data revealed similar improvements between the two treatments, with the advantage of fewer side effects associated with zoledronic acid [69,70,71]. In summary, parenteral bisphosphonates, with a focus on pamidronate and zoledronic acid, have undergone thorough investigation and showcased significant effectiveness in alleviating OA symptoms. Moreover, although their impact on managing skin manifestations is somewhat less pronounced than on OA symptoms, these treatments have still demonstrated a degree of effectiveness in this area. The available data collectively support the notion that the utilization of bisphosphonates is not only considered safe but also offers clear benefits for patients dealing with SAPHO syndrome.
Antibiotic treatment has been suggested to improve SAPHO symptoms, especially in acneiform lesions, and may partially be effective in OA findings. Antibiotics such as tetracycline, clindamycin, minocycline, and azithromycin have been used in various case series. In a prospective interventional study, 30 patients with roughly half having received prior DMARDs or biologics, were initiated on azithromycin. A loading dose of 500 mg was administered on six successive days, followed by 500 mg twice a week for 16 weeks. Among patients undergoing bone biopsy, approximately half exhibited positive cultures. By week 16, the antibiotics group demonstrated significant decreases in MRI scores, skin activity, osteitis activity, and health assessment scores. However, relapse occurred in patients within 12 weeks after discontinuing antibiotics [33]. Based on current data, antibiotic use may be appropriate in patients with moderate-to-severe acne vulgaris where skin involvement is prominent. Other treatment options should be considered for other involvements. As clinical data related to SAPHO increases over time, more detailed information may be available regarding the use of antibiotics in this disease [33, 34].
The utilization of biologics has emerged as a viable and effective alternative for SAPHO patients who do not respond to conventional treatments. This observation is largely drawn from multiple case reports, retrospective analyses of patient cohorts, and systematic reviews [72,73,74]. Among various biological treatment options, TNF blockers have gained prominence as the preferred choice due to their established track record [66, 74]. These agents have demonstrated efficacy in managing both OA manifestations and skin involvement. Notably, there is no clear superiority of one anti-TNF molecule over another in terms of therapeutic outcomes. The available evidence suggests that TNF blockers lead to significant improvement in about 90% of cases with OA involvement and approximately 70% in those with skin manifestations. However, it's important to acknowledge that there is a noteworthy observation concerning paradoxical skin reactions, including psoriasis, which can manifest in patients undergoing biologic treatments [63]. Therapies targeting IL-17, a pivotal cytokine in SAPHO disease pathogenesis arising from T cells, have demonstrated efficacy for both skin and OA manifestations. A systematic review revealed that among 5 patients treated with secukinumab, complete remission was observed in 2 (40%), and partial remission in one patient; most of these individuals were unresponsive to prior biological treatments [74]. In another study, a 24-week course of secukinumab (150 mg subcutaneous once weekly for 4 weeks and then every 4 weeks) resulted in substantial improvement in both OA and skin lesions for 4 patients, including one who was previously refractory to other biologics [73]. Ustekinumab (IL-12/23 inhibitor), another treatment targeting the Th-17 cytokine pathway, has limited experience, but a series of five patients suggested that 60% of OA symptoms and 55% of skin findings responded to this treatment [74, 75]. Ixekizumab also a humanized IgG4 monoclonal antibody that binds selectively to interleukin 17A was found to be successful in a patient with palmoplantar pustulosis [76]. Tildrakizumab is a humanized monoclonal antibody selectively targeting the p19 subunit of IL-23 used to successfully treat SAPHO in the case report [77].
In light of the autoinflammatory nature of SAPHO disease, anti-IL-1 treatments have been suggested to be a potential treatment option. A systematic review reported that improvement in OA symptoms was observed in 6 out of 7 patients (82.5%) receiving anti-IL-1 treatment. However, the review did not report a similarly good response for skin findings, with only 28.5% of patients experiencing improvement. Therefore, while anti-IL-1 treatments may be effective in managing OA symptoms, their effectiveness in treating skin involvement is limited [74].
JAK inhibitors have also emerged as a potential treatment option for SAPHO syndrome. A pilot study conducted retrospectively aimed to assess the efficacy of tofacitinib at a dosage of 5 mg twice daily in a group of 12 female patients. Notably, some of these patients had previously undergone treatment with anti-TNF agents and bisphosphonates [78]. The results of the study revealed promising outcomes, with significant improvements noted in OA symptoms among 83% of cases and skin manifestations showing improvement in 88% of cases. Additionally, there was a noticeable reduction in inflammatory changes observed on MRI, although to a slightly lesser extent. This suggests the potential value of JAK inhibitors, particularly tofacitinib, in addressing the diverse symptoms associated with SAPHO syndrome [78].
Tocilizumab's role in SAPHO treatment remains relatively limited, and its effectiveness has been questioned based on a small series involving two patients [79]. As of now, there is no available data on cases treated with rituximab for SAPHO syndrome.
Apremilast, a targeted sDMARD, functions by inhibiting the phosphodiesterase-4 enzyme expressed in leukocytes. In a notable case, a 24-year-old female who had previously undergone treatments involving various anti-TNFs, sekukinumab, and ustekinumab was prescribed apremilast—an oral PDE4 inhibitor. Interestingly, while the other treatments were accompanied by side effects, ustekinumab failed to effectively address OA symptoms. Consequently, apremilast was introduced as a replacement. The patient experienced positive outcomes, as both skin and bone symptoms displayed satisfactory improvements following the administration of apremilast [80].
In summary, the available data indicates that approximately 70% of SAPHO patients exhibit positive responses to treatment with NSAIDs and DMARDs. Similarly, a comparable response rate is observed with parenteral bisphosphonates. For those individuals who do not achieve desired outcomes with these initial treatments, biologics offer substantial benefits, with around 80% of patients experiencing improvements. Furthermore, in cases where the first biologic treatment proves ineffective, there is still potential for success, as about 70% of patients respond positively to a second biologic intervention [63,66,72, 74].
Difficulties in diagnosis and treatment
Diagnosing SAPHO disease poses a significant challenge due to its rarity and the presence of overlapping clinical features. On one hand, there are inflammation-related findings, and on the other, clinical features that resemble infections. Due to its rarity, clinicians may sometimes overlook to recognize this diverse disease spectrum. This complexity in clinical presentation often leads patients to seek care from different medical specialties. For instance, a patient with skin findings may consult a dermatologist, while someone experiencing back pain might be referred to neurosurgeons. Moreover, treatments administered by these specialists, such as corticosteroids, can potentially modify the disease's symptoms and course, complicating the process of arriving at the correct diagnosis. Furthermore, the varying degrees of disease severity add complexity to the diagnostic process, as milder forms may remain undetected.
In the management of SAPHO, the current situation is characterized by a shortage of controlled studies, and the available evidence mainly arises from case reports and expert opinions. Currently, there is no established treatment algorithm for this condition. Treatment decisions often revolve around targeting the dominant symptom area, whether it's related to skin disease, arthritis, or back pain. Consequently, treatment approaches are frequently borrowed from other inflammatory disorders, like psoriasis and inflammatory arthritis. Additionally, critical aspects such as the optimal treatment duration and the trajectory of the disease, including the probability of relapse post-treatment, remain largely unknown.
For diagnosing and treating SAPHO, a multidisciplinary approach is essential. Dermatologists are pivotal for assessing skin manifestations. Skin biopsies may be required. The treatment approach should be personalized, considering the patient's presentation, needs, comorbidities, symptoms, disease severity, and allergies.
Tips for clinicians
-
While SAPHO syndrome is rare, it's essential for clinicians to be aware of its existence and familiarize themselves with its clinical features.
-
Take a detailed medical history, including any previous skin conditions, joint pain, or bone problems. Ask about a family history of similar conditions, as some cases of SAPHO syndrome may have a genetic component.
-
Conduct a comprehensive physical examination, paying close attention to skin lesions, joint involvement, and areas of bone pain or swelling. Note any areas of tenderness or hyperostosis. Hyperostosis is excessive bone growth due to endosteal or periosteal reactions in the later stages of the disease.
-
For diagnosing and treating SAPHO, a multidisciplinary approach is essential. Dermatologists are pivotal for assessing skin manifestations. Skin biopsies may be required.
-
In treatment, specific drug classes have distinct roles and usages. For instance, antibiotics are primarily used for acneiform lesions in SAPHO and have limited effectiveness in treatment. Bisphosphonates are indicated for OA-dominant symptoms. NSAIDs are commonly paired with DMARDs, and corticosteroids serve as bridge therapy in specific cases. Intraarticular corticosteroids can be effective for localized disease. Some patients may necessitate combinations of DMARDs or biologic therapies. Therefore, treatment should be tailored to the individual's presentation, symptom severity, disease extent, and comorbidities.
Conclusion
In conclusion, SAPHO syndrome is a rare but important disease entity that poses diagnostic and therapeutic challenges to clinicians. It is a type of autoinflammatory rheumatic disease that shares similarities with SpA-group diseases. The lack of awareness and understanding of the disease can lead to significant diagnostic delays and inappropriate treatments. Thus, SAPHO syndrome should be considered in the differential diagnosis of patients presenting with skin and bone manifestations.
Future studies are needed to better understand the pathogenesis, clinical presentation, and optimal management of SAPHO syndrome. Large-scale prospective studies are necessary to establish the safety and efficacy of new and emerging targeted therapies. Furthermore, early diagnosis and multidisciplinary management are essential for achieving optimal outcomes in patients with SAPHO syndrome.
Data availability
Not applicable to this article as no datasets were generated during the current study.
References
Carneiro S, Sampaio-Barros PD (2013) SAPHO syndrome. Rheum Dis Clin North Am 39:401–418. https://doi.org/10.1016/J.RDC.2013.02.009
Chamot AMKM (1994) SAPHO syndrome. Z Rheumatol 53(4):234–242
Liu S, Tang M, Cao Y, Li C (2020) Synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome: review and update. Ther Adv Musculoskelet Dis. https://doi.org/10.1177/1759720X20912865
Nguyen MT, Borchers A, Selmi C et al (2012) The SAPHO syndrome. Semin Arthritis Rheum 42:254–265. https://doi.org/10.1016/J.SEMARTHRIT.2012.05.006
Chamot AM, Benhamou CL, Kahn MF, Beraneck L, Kaplan G, Prost A (1987) [Acne-pustulosis-hyperostosis-osteitis syndrome. Results of a national survey. 85 cases]. Rev Rhum Mal Osteoartic. 54(3):187–196
Hedrich CM, Morbach H, Reiser C, Girschick HJ (2020) New insights into adult and paediatric chronic non-bacterial osteomyelitis CNO. Curr Rheumatol Rep 22(9):52. https://doi.org/10.1007/S11926-020-00928-1
Kahn MF, Khan MA (1994) The SAPHO syndrome. Baillieres Clin Rheumatol 8:333–362. https://doi.org/10.1016/S0950-3579(94)80022-7
Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD (2011) Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int 31:1409–1417. https://doi.org/10.1007/S00296-011-1999-3
Przepiera-Bȩdzak H, Brzosko M (2021) SAPHO syndrome: pathogenesis, clinical presentation, imaging, comorbidities and treatment: a review. Adv Dermatol Allergol 38:937. https://doi.org/10.5114/ADA.2020.97394
Aljuhani F, Tournadre A, Tatar Z et al (2015) The SAPHO syndrome: a single-center study of 41 adult patients. J Rheumatol 42:329–334. https://doi.org/10.3899/JRHEUM.140342
Cao Y, Li C, Xu W et al (2019) Spinal and sacroiliac involvement in SAPHO syndrome: a single center study of a cohort of 354 patients. Semin Arthritis Rheum 48:990–996. https://doi.org/10.1016/J.SEMARTHRIT.2018.09.004
Colina M, Govoni M, Orzincolo C, Trotta F (2009) Clinical and radiologic evolution of synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome: a single center study of a cohort of 71 subjects. Arthritis Rheum 61:813–821. https://doi.org/10.1002/ART.24540
Hayem G, Bouchaud-Chabot A, Benali K et al (1999) SAPHO syndrome: a long-term follow-up study of 120 cases. Semin Arthritis Rheum 29:159–171. https://doi.org/10.1016/S0049-0172(99)80027-4
Berthelot JM, Corvec S, Hayem G (2018) SAPHO, autophagy, IL-1, FoxO1, and propionibacterium (Cutibacterium) acnes. Jt bone spine 85:171–176. https://doi.org/10.1016/J.JBSPIN.2017.04.010
Ferguson PJ, Lokuta MA, El-Shanti HI et al (2008) Neutrophil dysfunction in a family with a SAPHO syndrome-like phenotype. Arthritis Rheum 58:3264–3269. https://doi.org/10.1002/ART.23942
Gao S, Deng XL, Zhang L, Song L (2021) The comparison analysis of clinical and radiological features in SAPHO syndrome. Clin Rheumatol 40:349–357. https://doi.org/10.1007/S10067-020-05187-0
Rukavina I (2015) SAPHO syndrome: a review. J Child Orthop 9:19–27. https://doi.org/10.1007/S11832-014-0627-7
Assmann G, Wagner AD, Monika M et al (2010) Single-nucleotide polymorphisms p53 G72C and Mdm2 T309G in patients with psoriasis, psoriatic arthritis, and SAPHO syndrome. Rheumatol Int 30:1273–1276. https://doi.org/10.1007/S00296-009-1136-8
Hurtado-Nedelec M, Chollet-Martin S, Chapeton D et al (2010) Genetic susceptibility factors in a cohort of 38 patients with SAPHO syndrome: a study of PSTPIP2, NOD2, and LPIN2 genes. J Rheumatol 37:401–409. https://doi.org/10.3899/JRHEUM.090456
Zimmermann P, Curtis N (2019) The role of Cutibacterium acnes in auto-inflammatory bone disorders. Eur J Pediatr 178:89–95. https://doi.org/10.1007/s00431-018-3263-2
Rozin AP (2009) SAPHO syndrome: is a range of pathogen-associated rheumatic diseases extended? Arthritis Res Ther. https://doi.org/10.1186/AR2837
Firinu D, Barca MP, Lorrai MM et al (2014) TH17 cells are increased in the peripheral blood of patients with SAPHO syndrome. Autoimmunity 47:389–394. https://doi.org/10.3109/08916934.2014.906582
Vowels BR, Yang S, Leyden JJ (1995) Induction of proinflammatory cytokines by a soluble factor of Propionibacterium acnes: implications for chronic inflammatory acne. Infect Immun 63:3158–3165. https://doi.org/10.1128/IAI.63.8.3158-3165.1995
Dan Xu, Liu X, Chengyang Lu, Luo J, Wang C, Gao C, Jianfang Xie XL (2019) Reduction of peripheral natural killer cells in patients with SAPHO syndrome. Clin Exp Rheumatol 37(1):12–18
Trimble BS, Evers CJ, Ballaron SA, Young JM (1987) Intraarticular injection of Propionibacterium acnes causes an erosive arthritis in rats. Agents Actions 21:281–283. https://doi.org/10.1007/BF01966491
Zhang S, Li C, Zhang S et al (2019) Serum levels of proinflammatory, anti-inflammatory cytokines, and RANKL/OPG in synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome. Mod Rheumatol 29:523–530. https://doi.org/10.1080/14397595.2018.1469580
Lachmann HJ (2017) Periodic fever syndromes. Best Pract Res Clin Rheumatol 31:596–609. https://doi.org/10.1016/J.BERH.2017.12.001
Leuenberger M, Berner J, Di Lucca J et al (2016) PASS syndrome: an IL-1-Driven autoinflammatory disease. Dermatology 232:254–258. https://doi.org/10.1159/000443648
Sharma M, Ferguson PJ (2013) Autoinflammatory bone disorders: update on immunologic abnormalities and clues about possible triggers. Curr Opin Rheumatol 25:658–664. https://doi.org/10.1097/BOR.0B013E328363EB08
Zhao Y, Ferguson PJ (2018) Chronic nonbacterial osteomyelitis and chronic recurrent multifocal osteomyelitis in children. Pediatr Clin North Am 65:783–800. https://doi.org/10.1016/J.PCL.2018.04.003
Liao HJ, Chyuan IT, Wu CS et al (2015) Increased neutrophil infiltration, IL-1 production and a SAPHO syndrome-like phenotype in PSTPIP2-deficient mice. Rheumatology (Oxford) 54:1317–1326. https://doi.org/10.1093/RHEUMATOLOGY/KEU481
Hurtado-Nedelec M, Chollet-Nartin S, Nicaise-Roland P et al (2008) Characterization of the immune response in the synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO) syndrome. Rheumatology (Oxford) 47:1160–1167. https://doi.org/10.1093/RHEUMATOLOGY/KEN185
Assmann G, Kueck O, Kirchhoff T et al (2009) Efficacy of antibiotic therapy for SAPHO syndrome is lost after its discontinuation: an interventional study. Arthritis Res Ther 11(5):R140. https://doi.org/10.1186/AR2812
Colina M, Trotta F (2014) Antibiotics may be useful in the treatment of SAPHO syndrome. Mod Rheumatol 24:697–698. https://doi.org/10.3109/14397595.2013.874760
Kikuchi T, Fujii H, Fujita A et al (2018) Mandibular osteitis leading to the diagnosis of SAPHO syndrome. Case Rep Radiol 2018:1–6. https://doi.org/10.1155/2018/9142362
Cao Y, Li C, Yang Q et al (2019) Three patterns of osteoarticular involvement in SAPHO syndrome: a cluster analysis based on whole body bone scintigraphy of 157 patients. Rheumatology (Oxford) 58:1047–1055. https://doi.org/10.1093/RHEUMATOLOGY/KEY415
Li C, Ye Y, Cao Y et al (2020) Axial skeletal lesions and disease duration in SAPHO syndrome: a retrospective review of computed tomography findings in 81 patients. Int J Rheum Dis 23:1152–1158. https://doi.org/10.1111/1756-185X.13899
Wolber C, David-Jelinek K, Udvardi A et al (2011) Successful therapy of sacroiliitis in SAPHO syndrome by etanercept. Wien Med Wochenschr 161:204–208. https://doi.org/10.1007/S10354-010-0852-8
Van Doornum S, Barraclough D, McColl G, Wicks I (2000) SAPHO: rare or just not recognized? Semin Arthritis Rheum 30:70–77. https://doi.org/10.1053/SARH.2000.8371
Kotaki S, Gamoh S, Yoshida H et al (2020) SAPHO syndrome of the temporomandibular joint associated with trismus: a case report and review of the literature. Oral Radiol 36:197–202. https://doi.org/10.1007/S11282-019-00405-1
Orui H, Takahara M, Ishikawa A et al (2002) Radiological features of long bones in synovitis, acne, pustulosis, hyperostosis, osteitis syndrome and their correlation with pathological findings. Mod Rheumatol 12:56–63. https://doi.org/10.3109/S101650200009
Naves JE, Cabré E, Mañosa M et al (2013) A systematic review of SAPHO syndrome and inflammatory bowel disease association. Dig Dis Sci 58:2138–2147. https://doi.org/10.1007/S10620-013-2653-6
Carranco-Medina TE, Hidalgo-Calleja C, Calero-Paniagua I et al (2015) Thrombotic manifestations in SAPHO syndrome. Rev Lit Reumatol Clin 11:108–111. https://doi.org/10.1016/j.reuma.2014.07.003
Li C, Liu S, Sui X et al (2018) Pulmonary high-resolution computed tomography findings in patients with synovitis, acne, pustulosis, hyperostosis and osteitis syndrome. PLoS One. https://doi.org/10.1371/JOURNAL.PONE.0206858
Valentin R, Gürtler KF, Schaker A (1997) Renal amyloidosis and renal failure–a novel complication of the SAPHO syndrome. Nephrol Dial Transplant 12:2420–2423. https://doi.org/10.1093/NDT/12.11.2420
Heldmann F, Kiltz U, Baraliakos X, Braun J (2014) SAPHO syndrome. Z Rheumatol 73:729–741. https://doi.org/10.1007/S00393-014-1460-6
Baba Y, Weerakkody Y (2010) SAPHO syndrome. Radiopaedia.org, Melbourne. https://doi.org/10.53347/RID-8415
Sudoł-Szopińska I, Jans L, Teh J (2017) Rheumatoid arthritis: what do MRI and ultrasound show. J Ultrason 17:5. https://doi.org/10.15557/JOU.2017.0001
Xu T, Huang Y, Zhao Y et al (2022) 68Ga-DOTA-FAPI-04 PET/CT imaging in a case of SAPHO syndrome. Clin Nucl Med 47:246–248. https://doi.org/10.1097/RLU.0000000000003901
Przepiera-Będzak HBM (2016) Antiphospholipid syndrome with Antiβ2glicoprotein-1 antibodies as the cause of recurrent tibial vein thrombosis in SAPHO syndrome. Acta Dermatovenerol Croat 24(4):305306
Okuno H, Watanuki M, Kuwahara Y et al (2018) Clinical features and radiological findings of 67 patients with SAPHO syndrome. Mod Rheumatol 28:703–708. https://doi.org/10.1080/14397595.2017.1372874
Li C, Xiang Y, Wu X et al (2020) Serum IgG4 elevation in SAPHO syndrome: does it unmask a disease activity marker? Clin Exp Rheumatol 38:35–41
Fu Z, Liu M, Li Z et al (2016) Is the bullhead sign on bone scintigraphy really common in the patient with SAPHO syndrome? A single-center study of a 16-year experience. Nucl Med Commun 37:387–392. https://doi.org/10.1097/MNM.0000000000000451
Ferguson PJ, Sandu M (2012) Current understanding of the pathogenesis and management of chronic recurrent multifocal osteomyelitis. Curr Rheumatol Rep 14:130–141. https://doi.org/10.1007/S11926-012-0239-5
Schilling F, Kessler S (2000) SAPHO syndrome: clinico-rheumatologic and radiologic differentiation and classification of a patient sample of 86 cases. Z Rheumatol 59:1–28. https://doi.org/10.1007/S003930050001
Edwin J, Ahmed S, Verma S et al (2018) Swellings of the sternoclavicular joint: review of traumatic and non-traumatic pathologies. EFORT open Rev 3:471–484. https://doi.org/10.1302/2058-5241.3.170078
Rosenberg M, Conermann T (2022) Tietze syndrome. StatPearls, Treasure Island
Paice EW, Wright FW, Hill AGS (1983) Sternoclavicular erosions in polymyalgia rheumatica. Ann Rheum Dis 42:379–383. https://doi.org/10.1136/ARD.42.4.379
Carroll MB (2011) Sternocostoclavicular hyperostosis: a review. Ther Adv Musculoskelet Dis 3:101–110. https://doi.org/10.1177/1759720X11398333
Himuro H, Kurata S, Nagata S et al (2020) Imaging features in patients with SAPHO/CRMO: a pictorial review. Jpn J Radiol 38:622–629. https://doi.org/10.1007/S11604-020-00953-1
Jurik AG, Klicman RF, Simoni P et al (2018) SAPHO and CRMO: the Value of Imaging. Semin Musculoskelet Radiol 22:207–224. https://doi.org/10.1055/S-0038-1639469
Firinu D, Garcia-Larsen V, Manconi PE, Del Giacco SR (2016) SAPHO syndrome: current developments and approaches to clinical treatment. Curr Rheumatol Rep 18(6):35. https://doi.org/10.1007/S11926-016-0583-Y
Figueiredo ASB, Oliveira AL, Caetano A, Moraes-Fontes MF (2020) SAPHO: has the time come for tailored therapy? Clin Rheumatol 39:177–187. https://doi.org/10.1007/S10067-019-04675-2
Hussain A, Gondal M, Abdallah N et al (2020) Synovitis, acne, pustulosis, hyperostosis, osteitis (SAPHO): an interesting clinical syndrome. Cureus 12(9):e10184. https://doi.org/10.7759/CUREUS.10184
Jung J, Molinger M, Kohn D et al (2012) Intra-articular glucocorticosteroid injection into sternocostoclavicular joints in patients with SAPHO syndrome. Semin Arthritis Rheum 42:266–270. https://doi.org/10.1016/J.SEMARTHRIT.2012.03.012
Huang H, Zhang Z, Zhao J et al (2021) The effectiveness of treatments for patients with SAPHO syndrome: a follow-up study of 24 cases from a single center and review of literature. Clin Rheumatol 40:1131–1139. https://doi.org/10.1007/S10067-020-05322-X
Wang CR, Tsai YS, Whang-Peng J (2020) Zoledronic acid monotherapy improves osteoarticular involvement in SAPHO syndrome. Scand J Rheumatol 49:419–421. https://doi.org/10.1080/03009742.2020.1769179
Colina M, La Corte R, Trotta F (2009) Sustained remission of SAPHO syndrome with pamidronate: a follow-up of fourteen cases and a review of the literature. Clin Exp Rheumatol 27(1):112–115
Li C, Zhao Y, Zuo Y et al (2019) Efficacy of bisphosphonates in patients with synovitis, acne, pustulosis, hyperostosis, and osteitis syndrome: a prospective open study. Clin Exp Rheumatol 37(4):663–669
Liu S, Yin D, Lin Z et al (2023) Short-term efficacy of zoledronic acid in the treatment of 30 cases of SAPHO syndrome. Clin Exp Rheumatol. https://doi.org/10.55563/CLINEXPRHEUMATOL/ZPGYZ9
Zwaenepoel T, de Vlam K (2016) SAPHO: treatment options including bisphosphonates. Semin Arthritis Rheum 46:168–173. https://doi.org/10.1016/j.semarthrit.2016.04.004
Cheng W, Li F, Tian J et al (2022) New Insights in the treatment of SAPHO syndrome and medication recommendations. J Inflamm Res 15:2365. https://doi.org/10.2147/JIR.S353539
Wang L, Sun B, Li C (2021) Clinical and radiological remission of osteoarticular and cutaneous lesions in SAPHO patients treated with secukinumab: a case series. J Rheumatol 48:953–955. https://doi.org/10.3899/JRHEUM.201260
Daoussis D, Konstantopoulou G, Kraniotis P et al (2019) Biologics in SAPHO syndrome: a systematic review. Semin Arthritis Rheum 48:618–625. https://doi.org/10.1016/J.SEMARTHRIT.2018.04.003
Wendling D, Aubin F, Verhoeven F, Prati C (2017) IL-23/Th17 targeted therapies in SAPHO syndrome. A case series. Jt bone spine 84:733–735. https://doi.org/10.1016/J.JBSPIN.2017.05.016
Xia RY, Diao ZY, Zhang ZQ et al (2022) Successful treatment of synovitis, acne, pustulosis, hyperostosis and osteitis syndrome with ixekizumab. Clin Exp Dermatol 47:978–980. https://doi.org/10.1111/CED.15087
Licata G, Gambardella A, Calabrese G et al (2021) SAPHO syndrome successful treated with tildrakizumab. Dermatol Ther 34(1):e14758. https://doi.org/10.1111/DTH.14758
Li Y, Huo J, Cao Y et al (2020) Efficacy of tofacitinib in synovitis, acne, pustulosis, hyperostosis and osteitis syndrome: a pilot study with clinical and MRI evaluation. Ann Rheum Dis 79:1255–1257. https://doi.org/10.1136/ANNRHEUMDIS-2020-217250
Sun XC, Liu S, Li C et al (2018) Failure of tocilizumab in treating two patients with refractory SAPHO syndrome: a case report. J Int Med Res 46:5309–5315. https://doi.org/10.1177/0300060518806105
Adamo S, Nilsson J, Krebs A et al (2018) Successful treatment of SAPHO syndrome with apremilast. Br J Dermatol 179:959–962. https://doi.org/10.1111/BJD.16071
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
TDY and İS participated in the design of the study, carried out the literature search and selection process, and drafted the paper. All the authors participated in modelling the data, drafting the paper and reading and approving the final version of this manuscript. All authors take full responsibility for the integrity of all aspects of the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interests to declare.
Ethical approval
For this study ethical approval is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Demirci Yildirim, T., Sari, İ. SAPHO syndrome: current clinical, diagnostic and treatment approaches. Rheumatol Int (2023). https://doi.org/10.1007/s00296-023-05491-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00296-023-05491-3