
Abstract
Behçet’s disease (BD) is a variable vessel vasculitis characterized by recurrent oral and genital aphthosis accompanied by skin, ocular, gastrointestinal, neurologic, and articular involvement. BD is not common in childhood and the disease characteristics considerably differ between adults and children. 18 diagnostic/classification criteria have been published for BD to date. The pediatric BD (PEDBD) criteria, published in 2015, focused on pediatric BD, while the others mainly based on adult studies and are not validated for children. The aim of this review is to summarize the data about diagnostic/classification criteria for BD and to discuss the use and performance of the current criteria in pediatric BD. The covered topics are the characteristics of the diagnostic/classification criteria sets for BD, the factors restricting the universal use/acceptance of these criteria, and pediatric studies testing the performance of BD criteria sets. Having valid and universally accepted criteria with high performance is very important in pediatric BD as they help us determine patients for our studies and guide us through our clinical practice. There are less than 10 pediatric studies testing the performances of BD diagnostic/classification criteria. Their results suggest that revised ICBD (The International Criteria for BD) has the highest sensitivity, while ISG (The International Study Group) criteria remain as the most specific criteria set. Larger multinational pediatric BD cohorts with adequate control groups are required to compare the performance of the different criteria sets in children and to improve the performance of the existing PEDBD criteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Behçet’s disease (BD) is a variable vessel vasculitis affecting all sizes of vasculature in both the arterial and venous systems [1, 2]. Although it is seen all over the world, the main geographical distribution of BD is along the “Silk Road” (area between the Mediterranean region and Far East) [3]. BD is typically the disease of 20–40 years of age [4]. However, the disease is also complete in childhood in around 2.5–4.5% of all cases in recent cohorts [5,6,7]. The exact incidence/prevalence remains unknown, while the estimated prevalence of BD in children (< 15 years of age) was regarded as 1/600,000 according to a French nationwide survey [8]. Pediatric BD differs from juvenile-onset BD in the current terminology. If the disease is fully manifested and the diagnosis is made in childhood, this patient is considered to have pediatric BD [9,10,11]. On the other hand, in case of juvenile-onset BD, only the disease onset is in childhood (generally accepted as before 16 years of age) [12, 13]. That is, all pediatric BD patients have juvenile-onset BD, but not necessarily vice versa.
Behçet’s disease is characterized by recurrent oral and/or genital aphthous ulcers accompanied by cutaneous, ocular, articular, gastrointestinal, and/or central nervous system inflammatory lesions [2]. The most frequent manifestations are related to skin and ocular involvement [14]. It usually causes recurrent, self-limited disease flares like other autoinflammatory diseases [3]. The treatment is planned according to the spectrum of organ system involvement and disease severity. It ranges from colchicine (e.g., for mucocutaneous manifestations) to corticosteroids, immunosuppressive agents, and biologic drugs (e.g., for neurologic or arterial involvement) [14]. The prognosis depends on the site and severity of involvement. The leading causes of BD-related morbidity and mortality are ocular, neurologic, and arterial involvements [15].
Up to date, 18 diagnostic/classification criteria sets have been published for BD [11, 16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35]. No other primary systemic vasculitis has ever had that many diagnostic/classification criteria. The main reason for this is probably the diversity of the disease phenotype among patients, mainly due to different ethnicity and/or country of residence. Differences in the clinical features among different age groups and the changing clinical picture of BD over time [such as the gradual decline in the frequency of pathergy test (PT) positivity in several countries [36]] also contribute to the need for frequent modifications in the diagnostic/classification criteria for BD.
In 1946, Curth developed the first criteria set for BD [25]. Until 1990, eight more criteria sets had been developed [18,19,20,21, 26,27,28,29,30]. All but one were reported from different countries (one each from France [18, 19], UK [20], USA [28], China [29], and Turkey [30]; and two from Japan as the original [26] and revised forms [21] of the same criteria set). The criteria set by Hubault&Hamza was developed as a result of a collaboration between France and Tunisia [27]. In 1990 and 1992, the International Study Group (ISG) criteria set was presented based on a study including 912 patients with BD from 12 centers of seven different countries [23, 24]. It has been the most widely used criteria set since then. Between 1992 and 2004, four criteria sets (Iran traditional criteria [31], Iran classification tree [34], revised Dilsen [33], and Korea criteria [16, 17]) had been developed. The International Team for the revision of the ISG criteria was formed in 2004 and the International Criteria for BD (ICBD) was developed in 2006 by this team [32]. The original ICBD had both traditional and classification tree forms. ICBD was revised in 2010 [22]. In 2015, consensus classification criteria set was created for pediatric BD (PEDBD) for the first time [11]. PEDBD criteria set was formed based on a cohort of 219 children with BD from 42 centers in 12 countries [11].
The BD criteria are helpful for disease diagnosis especially for physicians who are not BD experts. It is also possible to compare the results of different studies and conduct multicenter/multinational studies efficiently with a universally accepted criteria set [37].
BD is a heterogeneous vasculitis and the disease characteristics differ according to the age at onset, gender, ethnicity, and country of residence [3]. These differences restrict acceptance and use of different criteria sets universally. Especially pediatric and adult BD patients differ considerably with regard to clinical characteristics, which makes it difficult to use the same diagnostic/classification criteria set for both patient groups.
The aim of this review is to provide an overview of the current data about diagnostic/classification criteria for BD, analyze the factors restricting the universal use of them, and investigate their use and performances in pediatric BD patients.
Search strategy
This review was conducted according to the guidance on narrative reviews [38]. The Cochrane Library and MEDLINE/PubMed databases were searched from database inception to October 1, 2018 using the following keywords: (“Behçet’s disease” OR “Adamantiades-Behçet disease” OR “Behçet syndrome”) AND (“diagnosis” OR “classification” OR “criteria”). Case series, original research articles, and review articles with a focus on diagnostic/classification criteria for BD were analyzed. The search was primarily focused on pediatric BD studies. However, all relevant adult studies and studies discovered from references of the analyzed articles were also evaluated. The search was restricted to English articles.
Different criteria sets for diagnosis/classification of BD
BD manifestations in different criteria sets are summarized in Table 1.
The disease characteristics were divided into major and minor manifestations in Hewitt [18], Mason&Barnes [20], Hubault&Hamza [27], Cheng and Zhang [29], Dilsen [30], and revised Japan criteria [21]. Oral aphthosis (OA), genital aphthosis (GA), and ocular manifestations (OM) were classified as major criteria in all aforementioned criteria sets with the addition of skin manifestations (SM) in Mason and Barnes [20], Dilsen [30], and revised Japan [21], and thrombophlebitis in Dilsen criteria [30]. In addition to the major/minor discrimination, higher values were attributed to certain items in some of the aforementioned criteria sets: OM in Hewitt criteria [18] and PT in Dilsen [30] and Hubault&Hamza criteria [27]. In addition, note that, in Dilsen criteria, PT was distinguished from major/minor discrimination [30].
In the rest of the criteria sets, there was no separation as major/minor. However, the value of all items was not equal in most of them. The higher valued items were OM in Japan [26] and Iran criteria [31], OA and GA in O’Duffy criteria [28], GA in Korea criteria [16, 17], OA in the ISG criteria [23, 24], OM and GA in ICBD [32], and OA, OM, and GA in revised ICBD [22].
Among all criteria sets, the most frequently higher valued item was OM followed by GA and OA. A mandatory item (OA) is only present in the ISG criteria among all criteria sets [23, 24].
All items had equal value in Curth [25], revised Dilsen [33], and PEDBD criteria [11].
The ISG, ICBD, and PEDBD criteria sets are discussed below in detail.
Factors restricting the universal use of diagnostic/classification criteria for BD
There are several factors mainly based on the heterogeneity of the disease restricting the universal use of diagnostic/classification criteria sets for BD among all patients:
-
1.
The disease phenotype differs among different age groups especially between children and adults.
-
2.
The frequency of organ manifestations is different among females and males.
-
3.
The disease characteristics differ according to the ethnicity and the country of residence.
-
4.
The clinical practice for BD differs among different countries or even different health care centers.
Differences between adult and pediatric cases with BD
There are several differences between adults and children with BD. The disease characteristics in large pediatric and adult BD cohorts are presented in Table 2.
First of all, there is a longer period between the onset of the first symptom and full-blown disease in children than adults [9, 39]. In addition, in more than 80% of BD patients, the disease is not complete before the age of 16 years [40]. Thus, children do not fulfill some BD diagnostic/classification criteria for a longer time than adults, although they have the disease.
Other differences include more frequent neurologic involvement, gastrointestinal involvement, and family history of BD and less frequent ocular manifestations in children than adults [5, 7, 11,12,13, 22, 41,42,43]. Furthermore, perianal aphthosis seems to be a specific feature of pediatric BD [44]. The higher frequency of family history points at a more redundant genetic load which may modify the disease phenotype. Of note, especially in patients with a positive family history, we should keep in mind the recently defined monogenic disease, haploinsufficiency of A20 (HA20) which could mimic early-onset BD [45, 46].
Another issue is the modifying effect of puberty on BD phenotype. GA is less common in pre-pubertal BD patients [44]. Beside GA, sex hormones probably affect PT positivity [47].
As a result of all aforementioned factors, pediatric BD differs from adult BD which makes it difficult to use the same diagnostic/classification criteria set for both. Consequently, applying the criteria sets based on adult studies may restrict early diagnosis of BD in children and inclusion of accurate patient groups to studies.
Differences among patients according to gender
A meta-analysis on gender-specific differences in BD (both adult and pediatric) revealed that ocular involvement, folliculitis, papulopustular lesions, vascular involvement, superficial/deep vein thrombosis, and positive PT were associated with male gender, while GA, joint involvement, and erythema nodosum were associated with female gender [48]. It is important to note that the study results suggested a more severe disease in males than females [48,49,50].
In pediatric BD, uveitis is more common and severe in boys than girls in most of the series [44, 51,52,53]. In the largest international pediatric BD cohort (156 patients from 12 different countries), Koné-Paut et al. demonstrated that cutaneous, ocular, and vascular manifestations were associated with male gender, while GA was associated with female gender [11]. The results of the recent Italian pediatric BD cohort study (n = 110) were consistent with these findings [42]. In addition, similar results have been reported from the most recent large Iranian cohort (n = 204), where GA was more frequent in girls and OM (especially severe forms such as posterior uveitis, retinal vasculitis, and panophthalmitis), erythema nodosum, and monoarthritis were more prevalent in boys [7].
Differences between BD patients according to the ethnicity/country of residence
The frequency of different BD manifestations differs according to the ethnicity and the country of residence as well. This further restricts the universal applicability of diagnostic/classification criteria sets and may be one of the reasons for different performance of the same criteria set in patient groups from different countries.
An increasing number of adult BD studies are pointing at differences in disease characteristics associated with different ethnicity and/or country of residence. Sibley et al. showed that BD patients from Turkey were more likely to have gastrointestinal and neurologic involvement when compared to patients from USA [54]. In addition, Kobayashi et al. demonstrated that GA was less frequent, while epididymitis and pulmonary involvement were more frequent among Japanese patients than American patients [55]. Another recent study by Moosmann et al. has emphasized the effect of the country of residence on disease characteristics [56]. They have shown that neurological, gastrointestinal, and vascular involvements were less frequent, while GA and SM were more frequent in Turkish BD patients living in Austria compared to those living in Turkey [56]. Other than clinical differences, human leukocyte antigen B51 (HLA-B51) and PT positivity differ among different countries. HLA-B51 positivity is more frequent in patients from Japan and Mediterranean countries when compared to those from Northern Europe or USA [44]. The PT positivity is similarly higher in the “Silk Road” countries [57].
The differences are less evident in pediatric BD, since there is a lack of direct comparison studies with large number of patients. In the international pediatric BD cohort published in 1998, neurologic and gastrointestinal involvement were more frequent in patients from France and Saudi Arabia, while SM was more common in patients from Turkey [44]. Nanthapisal et al. reported that gastrointestinal and neurologic manifestations were more frequent in UK pediatric BD cohort when compared to non-UK ones [10]. In our previous study including pediatric BD patients from Turkey and Israel, neurologic involvement was more frequent among patients from Turkey [58]. In the international cohort evaluated during the development of PEDBD criteria (n = 156), patients were younger at disease onset and skin and vascular involvements were less frequent in European than non-European children [11]. In the most recent and largest pediatric BD series from Iran (n = 204), GA, SM (especially erythema nodosum), and gastrointestinal involvement were less frequent, while ocular lesions were more frequent and severe compared to other cohorts [7].
Differences in clinical practice for BD
Almost all of the items included in the diagnostic/classification criteria sets of BD are clinical manifestations that the physicians directly observe or get information about from the patient. An exception to this is PT, which is a nonspecific cutaneous hypersensitivity reaction to a prick skin trauma [59]. It is positive in 37–80% of pediatric patients [12, 44]. Since the rate of positivity varies widely in different populations, this test is not routinely performed in all countries. Davatchi et al. studied the impact of the positive PT on the performance of diagnostic/classification criteria for BD [60]. They found that the majority of the criteria sets lost sensitivity, but gained specificity when the PT was excluded [60]. The PT is excluded from the PEDBD criteria, since there is a large variation of its presence in the pediatric population [12, 44]. It is an optional item in the revised ICBD, since it is not performed routinely everywhere [22]. According to the revised ICBD, if the PT is performed and the patient gets a positive reaction, it counts. Otherwise, it does not affect the performance of the criteria set. Thus, it is not a must to perform this test for BD diagnosis.
To overcome the obstacles originating from the heterogeneity of the disease and clinical practice, it is important to have diagnostic/classification criteria based on large international cohorts of patients from all age groups while focusing on common clinical practice. This has mostly been achieved in the revised ICBD, where more than 2500 patients from 27 different countries were included under the expertise of scientists from 32 countries [22]. However, pediatric BD was not specifically addressed in this criteria set.
There are major differences between pediatric and adult BD. Thus, using different criteria sets could help with the early diagnosis especially in pediatric patients. Having said that, the sensitivity of the revised ICBD was higher than the sensitivity of PEDBD criteria in two recent studies testing both criteria sets in children with BD [7, 42].
Comparing ISG, and ICBD, and PEDBD criteria in pediatric BD patients
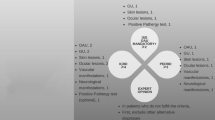
The Venn diagram of ISG, revised ICBD, and PEDBD criteria sets is presented in Fig. 1. In addition, studies (with ≥ 30 BD patients) testing the performance of BD diagnostic/classification criteria in pediatric BD are presented in Table 3.

Venn diagram of ISG (the International Study Group), revised ICBD (the International Criteria for Behçet’s disease), and PEDBD (pediatric Behçet’s disease) criteria sets with Behçet’s disease definitions indicated for each one along with their items. GA genital aphthosis, NM neurologic manifestations, OA oral aphthosis, OM ocular manifestations, PPT positive pathergy test, SM skin manifestations, VM vascular manifestations
The ISG criteria set was presented in 1990 and 1992 [23, 24]. According to the ISG criteria, OA is mandatory. In addition, the patient should have two of the following four features to be classified as having BD: GA, SM (pseudofolliculitis and erythema nodosum), OM, and positive PT.
The ICBD was created in 2006 and revised in 2010 [22, 32]. The original ICBD has a traditional format and a classification tree [32]. According to the traditional format, the patient gets two points for each GA and OM and one point for each OA, SM (pseudofolliculitis, erythema nodosum, and skin aphthosis), vascular manifestations (VM) (arterial and venous), and positive PT. A patient is classified as having BD if she/he gets 3 or more points. There are five scenarios in the classification tree as follows: OA + GA, OA + OM, GA + OM, OM + VM, and OA + SM + positive PT. Patients with one of these subsets are classified with BD. There are three changes in the revised ICDB. First, a new criterion is added as neurologic manifestations, which is worth one point. Second, the value of OA is changed as two points. Third, a patient should have 4 or more points to be classified as having BD [22]. Of note, in the revised ICBD, positive PT was an optional criterion.
In 2015, an international expert consensus group (PEDBD group) suggested new classification criteria for pediatric BD based on a large pediatric cohort including mainly European patients with the supplementation of Turkish patients [11]. In the PEDBD criteria set, all symptom categories have the same weight, the PT is not included, and OA is not a mandatory criterion. For classifying a patient as having BD, the patient should have three or more of the following criteria: OA (≥ 3 attacks per year), GA (typical with scars), SM (necrotic folliculitis, acneiform lesions, and erythema nodosum), neurologic involvement (except isolated headaches), OM (anterior uveitis, posterior uveitis, and retinal vasculitis), and VM (venous thrombosis, arterial thrombosis, and arterial aneurysms).
Main differences of the ISG criteria from ICBD and PEDBD criteria sets are as follows: OA is a mandatory criterion and neurologic and vascular manifestations are not included in the ISG criteria set. We are aware that there are BD cases without OA [61]. Thus, inclusion of OA as a mandatory criterion was probably one of the factors limiting the performance of the ISG criteria. In an international collaborative study, 21 out of 86 pediatric BD cases did not fulfill the ISG criteria [44]. GA, SM, hypersensitivity, and uveitis were less frequent, whereas neurologic symptoms were more frequent in these patients compared to the ones fulfilling the ISG criteria [44]. In another study, expert pediatric rheumatologists observed that almost half of the definitive or probable BD patients did not fulfill the ISG criteria [52]. Nanthapisal et al. reported that revised ICBD had an almost 4 times higher sensitivity than the ISG criteria in the UK-based pediatric BD cohort [10]. In the original report of PEDBD criteria, the sensitivity of PEDBD was higher, while specificity was lower than the ISG criteria (Table 3) [11]. In the first study comparing the performances of the ISG and PEDBD criteria in children after PEDBD publication, it was demonstrated that the sensitivity of PEDBD criteria was higher than the ISG criteria, while the specificities were quite similar (Table 3) [58]. In the recent Italian cohort (n = 110), the sensitivity of PEDBD was higher than the ISG criteria, while vice versa was true in the Iranian cohort (n = 204) (Table 3) [7, 42].
The items of the revised ICBD and PEDBD criteria sets are quite similar. Only difference is the inclusion of positive PT as an optional item in ICBD when it is absent in PEDBD criteria. However, the values of the items and the prerequisites for classifying a patient with BD differ as mentioned above. There is only one scenario, where PEDBD classifies a patient with BD and revised ICBD does not. It is when the patient has only the trio of BD-related SM–VM–neurologic involvement. On the contrary, there are multiple possible scenarios that the revised ICBD classifies a patient with BD and PEDBD does not. For instance, OA + GA, OA + OM, GA + OM, OA + GA + positive PT, OA + OM + positive PT, GA + OM + positive PT, OA + SM + positive PT, and OA + VM + positive PT can be given as such scenarios. This is important especially for patients with OA + GA and OA + OM, since these scenarios are quite common among pediatric BD patients unlike the ones including positive PT. Two studies including large cohorts of pediatric BD patients (n = 110 in the Italian cohort and n = 204 in the Iranian cohort) have compared the performance of ISG, revised ICBD, and PEDBD criteria and they found that the revised ICBD had the highest sensitivity (Table 3) [7, 42]. The difference between values of items and prerequisites is probably one of the factors affecting the lower sensitivity of the PEDBD criteria. Another possible contributing factor is the exclusion of PT. Including positive PT as a part of SM to the PEDBD criteria set could improve the sensitivity [7].
Although OA is not seen in all BD patients, it is by far the most common clinical manifestation of BD, both in adults and children [4, 7]. Giving a higher value to OA item could improve the performance of the criteria sets. This may be one reason for revised ICBD having a higher sensitivity than PEDBD. Of note, defining the recurrence of BD-related OA may be important to keep the specificity of the criteria set at a desired level. The recurrence of OA is defined as at least 3 attacks per year in PEDBD and ISG criteria [11, 23, 24].
The definition of single items is also important and some definitions could affect the performance of the criteria sets. GA was defined as being “typically with scars” in PEDBD criteria [11]. However, a previous study demonstrated that nearly, one-third of BD genital ulcers did not heal with scarring [62]. Acneiform lesions were defined as being “observed by a physician in postadolescent patients not on corticosteroid treatment” in the ISG criteria [23, 24]. We now know that acneiform lesions appear in around 10% of pediatric BD patients [42, 58] and this is included in the SM criterion of PEDBD [11]. Of note, acneiform lesions were not mentioned in the revised ICBD [22]. However, skin aphthosis was only mentioned in the revised ICBD.
Conclusion
It is tempting to have one valid diagnostic/classification criteria set with considerably high performance in both adult and pediatric BD patients and among all countries. However, as discussed above, there are prominent differences among different groups of patients. At least, it seems reasonable to have two different criteria sets; one for children and one for adults. Another option is to use different versions of the same criteria set. The values of different items could be changed when the revised ICBD is being used for pre-pubertal children. To be able to revise criteria sets properly to fit both age groups, large cohort studies including both children and adults are required.
Finally, it is important to emphasize that no diagnostic/classification criteria will have 100% sensitivity and specificity among all patients with BD unless a pathognomonic feature is found. Although we get guidance from these criteria, we should decide on the diagnosis of the patient on an individual basis. After all, the golden standard for diagnosis is the expert opinion in almost all of the studies that reported a diagnostic/classification criteria set for BD. In addition, BD is one of the most likely diagnosis in a child with recurrent OA, pulmonary arterial aneurysm, and deep vein thrombosis along with high acute phase reactants, even though this patient is not classified with BD by any of the ISG ICBD, or PEDBD criteria sets.
References
Batu ED, Ozen S (2012) Pediatric vasculitis. Curr Rheumatol Rep 14(2):121–129. https://doi.org/10.1007/s11926-011-0232-4
Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA (2013) 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum 65(1):1–11. https://doi.org/10.1002/art.37715
Kone-Paut I (2016) Behcet’s disease in children, an overview. Pediatr Rheumatol 14(1):10. https://doi.org/10.1186/s12969-016-0070-z
Davatchi F, Chams-Davatchi C, Shams H, Shahram F, Nadji A, Akhlaghi M, Faezi T, Ghodsi Z, Sadeghi Abdollahi B, Ashofteh F, Mohtasham N, Kavosi H, Masoumi M (2017) Behcet’s disease: epidemiology, clinical manifestations, and diagnosis. Expert Rev Clin Immunol 13(1):57–65. https://doi.org/10.1080/1744666X.2016.1205486
Atmaca L, Boyvat A, Yalcindag FN, Atmaca-Sonmez P, Gurler A (2011) Behcet disease in children. Ocul Immunol Inflamm 19(2):103–107. https://doi.org/10.3109/09273948.2011.555592
Ishido T, Horita N, Takeuchi M, Kawagoe T, Shibuya E, Yamane T, Hayashi T, Meguro A, Ishido M, Minegishi K, Yoshimi R, Kirino Y, Kato S, Arimoto J, Ishigatsubo Y, Takeno M, Kurosawa M, Kaneko T, Mizuki N (2017) Clinical manifestations of Behcet’s disease depending on sex and age: results from Japanese nationwide registration. Rheumatology 56(11):1918–1927. https://doi.org/10.1093/rheumatology/kex285
Shahram F, Nadji A, Akhlaghi M, Faezi ST, Chams-Davatchi C, Shams H, Ghodsi SZ, Davatchi F (2018) Paediatric Behcet’s disease in Iran: report of 204 cases. Clin Exp Rheumatol 36
Koné-Paut IBJ (1993) La maladie de Behçet chez l’enfant en France. Arch Fr Pediatr 50:561–563
Borlu M, Uksal U, Ferahbas A, Evereklioglu C (2006) Clinical features of Behcet’s disease in children. Int J Dermatol 45(6):713–716. https://doi.org/10.1111/j.1365-4632.2006.02754.x
Nanthapisal S, Klein NJ, Ambrose N, Eleftheriou D, Brogan PA (2016) Paediatric Behcet’s disease: a UK tertiary centre experience. Clin Rheumatol 35(10):2509–2516. https://doi.org/10.1007/s10067-016-3187-z
Kone-Paut I, Shahram F, Darce-Bello M, Cantarini L, Cimaz R, Gattorno M, Anton J, Hofer M, Chkirate B, Bouayed K, Tugal-Tutkun I, Kuemmerle-Deschner J, Agostini H, Federici S, Arnoux A, Piedvache C, Ozen S, Group P (2015) Consensus classification criteria for paediatric Behcet’s disease from a prospective observational cohort: PEDBD. Ann Rheum Dis 75(6):958–964. https://doi.org/10.1136/annrheumdis-2015-208491
Karincaoglu Y, Borlu M, Toker SC, Akman A, Onder M, Gunasti S, Usta A, Kandi B, Durusoy C, Seyhan M, Utas S, Saricaoglu H, Ozden MG, Uzun S, Tursen U, Cicek D, Donmez L, Alpsoy E (2008) Demographic and clinical properties of juvenile-onset Behcet’s disease: a controlled multicenter study. J Am Acad Dermatol 58(4):579–584. https://doi.org/10.1016/j.jaad.2007.10.452
Kitaichi N, Ohno S (2008) Behcet disease in children. Int Ophthalmol Clin 48(3):87–91. https://doi.org/10.1097/IIO.0b013e31817d8393
Davatchi F (2014) Behcet’s disease. Int J Rheum Dis 17(4):355–357. https://doi.org/10.1111/1756-185X.12378
Saadoun D, Wechsler B (2012) Behcet’s disease. Orphanet J Rare Dis 7:20. https://doi.org/10.1186/1750-1172-7-20
Chang HK, Kim SY (2003) Survey and validation of the criteria for Behcet’s disease recently used in Korea: a suggestion for modification of the International Study Group criteria. J Korean Med Sci 18(1):88–92. https://doi.org/10.3346/jkms.2003.18.1.88
Chang HK, Lee SS, Bai HJ, Lee YW, Yoon BY, Lee CH, Lee YH, Song GG, Chung WT, Lee SW, Choe JY, Kim CG, Chang DK (2004) Validation of the classification criteria commonly used in Korea and a modified set of preliminary criteria for Behcet’s disease: a multi-center study. Clin Exp Rheumatol 22(4 Suppl 34):S21–S26
Hewitt J, Escande JP, Lauret P, Perlemuter L (1969) Criteria for diagnosis of Behcet’s syndrome. Bull Soc Fr Dermatol Syphiligr 76(4):565–568
Hewitt J, Escande JP, Manesse S (1971) Revision of the diagnostic criteria of Behcet’s syndrome. Presse Med 79(20):901
Mason RM, Barnes CG (1969) Behcet’s syndrome with arthritis. Ann Rheum Dis 28(2):95–103
Mizushima Y (1988) Recent research into Behcet’s disease in Japan. Int J Tissue React 10(2):59–65
International Team for the Revision of the International Criteria for Behcet’s D (2014) The International Criteria for Behcet’s Disease (ICBD): a collaborative study of 27 countries on the sensitivity and specificity of the new criteria. J EurAcad Dermatol Venereol 28(3):338–347. https://doi.org/10.1111/jdv.12107
International Study Group for Behcet’s Disease (1990) Criteria for diagnosis of Behcet’s disease. Lancet 335(8697):1078–1080
The International Study Group for Behcet’s disease (1992) Evaluation of diagnostic (‘classification’) criteria in Behcet’s disease–towards internationally agreed criteria. Br J Rheumatol 31(5):299–308
Curth HO (1946) Recurrent genito-oral aphthosis with hypopion (Behcet’s syndrome). Arch Dermatol 54:179–196
Behcet’s Disease Research Committee of Japan (1974) Behcet’s disease guide to the diagnosis of Behcet’s disease. Japan J Ophthalmol 18:291–294
Hubault A, Hamza M (1974) La maladie de Behçet en 1974. In: de Seze S et al (eds) L’actualite rhumatologique, vol 15. Expension Scientifique, Paris, pp 43–55
O’Duffy JD (1974) Critères proposés pour le diagnostique de la maladie de Behçet et notes therapeutiques. Rev Med (Paris) 36:2371–2379
Cheng SP, Zhang X-Q (1980) Some special clinical manifestations of Behçet’s disease-report of illustrative cases and review of the literature (author’s transl). Chinese J Intern Med 19:15–22
Dilsen N, Konice M, Aral O (1986) Our diagnostic criteria of Behcet’s disease-an overview. In: Lehner T, Barnes CG (eds) Recent advances in Behçet’s disease, vol International Congress and Symposium Series 103. London Royal Society of Medicine Services, London, pp 177–180
Davatchi F, Shahram F, Akbarian M et al (1993) Accuracy of existing diagnostic criteria for Behçet’s disease. In: Wechsler B, Godeau P (eds) Behcet’s disease. Excerpta Medica International Congress Series 1037, Amsterdam, pp 225–228
International Team for the Revision of the International Criteria for Behçet’s disease (2006) Revision of the International Criteria for Behçet’s disease (ICBD). Clin Exp Rheumatol 24(Suppl. 42):S14–S15
Dilsen N (2000) About diagnostic criteria for Behcet’s Disease: our new proposal. In: Bang D, Lee ES, Lee S (eds) Behçet’s Disease. Design Mecca Publishing Co., Seoul, pp 101–104
Davatchi F, Shahram F, Akbarian M et al (1993) Classification tree for the diagnosis of Behçet’s disease. In: Wechsler B, Godeau P (eds) Behçet’s Disease. Excerpta Medica International Congress Series 1037, Amsterdam, pp 245–248
Davatchi F, Sadeghi Abdollahi B, Chams-Davatchi C, Shahram F, Shams H, Nadji A, Faezi T, Akhlaghi M, Ghodsi Z, Mohtasham N, Ashofteh F (2015) The saga of diagnostic/classification criteria in Behcet’s disease. Int J Rheum Dis 18(6):594–605. https://doi.org/10.1111/1756-185X.12520
Davatchi F, Chams-Davatchi C, Shahram F, Nadji A, Shams H, Ghodsi Z, Akhlaghi M, Naderi N, Sadeghi-Abdolahi B (2007) Pathergy test in Behçet’s disease: change in incidence over the time. APLAR J Rheumatol 10:333–335
Batu ED, Ozen S (2015) Vasculitis: do we know more to classify better? Pediatr Nephrol 30(9):1425–1432. https://doi.org/10.1007/s00467-014-3015-0
Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD (2011) Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int 31(11):1409–1417. https://doi.org/10.1007/s00296-011-1999-3
Treudler R, Orfanos CE, Zouboulis CC (1999) Twenty-eight cases of juvenile-onset Adamantiades-Behcet disease in Germany. Dermatology 199(1):15–19. https://doi.org/10.1159/000018197
Kotter I, Vonthein R, Muller CA, Gunaydin I, Zierhut M, Stubiger N (2004) Behcet’s disease in patients of German and Turkish origin living in Germany: a comparative analysis. J Rheumatol 31(1):133–139
Davatchi F, Chams-Davatchi C, Shams H, Nadji A, Faezi T, Akhlaghi M, Sadeghi Abdollahi B, Ashofteh F, Ghodsi Z, Mohtasham N, Shahram F (2016) Adult Behcet’s disease in Iran: analysis of 6075 patients. Int J Rheum Dis 19(1):95–103. https://doi.org/10.1111/1756-185X.12691
Gallizzi R, Pidone C, Cantarini L, Finetti M, Cattalini M, Filocamo G, Insalaco A, Rigante D, Consolini R, Maggio MC, Civino A, Martino S, Olivieri AN, Fabio G, Pastore S, Mauro A, Sutera D, Trimarchi G, Ruperto N, Gattorno M, Cimaz R (2017) A national cohort study on pediatric Behcet’s disease: cross-sectional data from an Italian registry. Pediatr Rheumatol Online J 15(1):84. https://doi.org/10.1186/s12969-017-0213-x
Kim DY, Choi MJ, Cho S, Kim DW, Bang D (2014) Changing clinical expression of Behcet disease in Korea during three decades (1983–2012): chronological analysis of 3674 hospital-based patients. Br J Dermatol 170(2):458–461. https://doi.org/10.1111/bjd.12661
Kone-Paut I, Yurdakul S, Bahabri SA, Shafae N, Ozen S, Ozdogan H, Bernard JL (1998) Clinical features of Behcet’s disease in children: an international collaborative study of 86 cases. J Pediatr 132(4):721–725
Aeschlimann FA, Batu ED, Canna SW, Go E, Gul A, Hoffmann P, Leavis HL, Ozen S, Schwartz DM, Stone DL, van Royen-Kerkof A, Kastner DL, Aksentijevich I, Laxer RM (2018) A20 haploinsufficiency (HA20): clinical phenotypes and disease course of patients with a newly recognised NF-kB-mediated autoinflammatory disease. Ann Rheum Dis 77(5):728–735. https://doi.org/10.1136/annrheumdis-2017-212403
Ozen S, Batu ED (2018) Vasculitis Pathogenesis: can we talk about precision medicine? Front Immunol 9:1892. https://doi.org/10.3389/fimmu.2018.01892
Alpsoy E, Elpek GO, Yilmaz F, Ciftcioglu MA, Akman A, Uzun S, Karakuzu A (2005) Androgen receptor levels of oral and genital ulcers and skin pathergy test in patients with Behcet’s disease. Dermatology 210(1):31–35. https://doi.org/10.1159/000081480
Bonitsis NG, Luong Nguyen LB, LaValley MP, Papoutsis N, Altenburg A, Kotter I, Micheli C, Maldini C, Mahr A, Zouboulis CC (2015) Gender-specific differences in Adamantiades-Behcet’s disease manifestations: an analysis of the German registry and meta-analysis of data from the literature. Rheumatology 54(1):121–133. https://doi.org/10.1093/rheumatology/keu247
Kural-Seyahi E, Fresko I, Seyahi N, Ozyazgan Y, Mat C, Hamuryudan V, Yurdakul S, Yazici H (2003) The long-term mortality and morbidity of Behcet syndrome: a 2-decade outcome survey of 387 patients followed at a dedicated center. Medicine 82(1):60–76
Saadoun D, Wechsler B, Desseaux K, Le Thi Huong D, Amoura Z, Resche-Rigon M, Cacoub P (2010) Mortality in Behcet’s disease. Arthritis Rheum 62(9):2806–2812. https://doi.org/10.1002/art.27568
Kim DK, Chang SN, Bang D, Lee ES, Lee S (1994) Clinical analysis of 40 cases of childhood-onset Behcet’s disease. Pediatr Dermatol 11(2):95–101
Kone-Paut I, Darce-Bello M, Shahram F, Gattorno M, Cimaz R, Ozen S, Cantarini L, Tugal-Tutktun I, Assaad-Khalil S, Hofer M, Kuemmerle-Deschner J, Benamour S, Al Mayouf S, Pajot C, Anton J, Faye A, Bono W, Nielsen S, Letierce A, Tran TA, Committee P-BIE (2011) Registries in rheumatological and musculoskeletal conditions. Paediatric Behcet’s disease: an international cohort study of 110 patients. One-year follow-up data. Rheumatology 50(1):184–188. https://doi.org/10.1093/rheumatology/keq324
Krause I, Uziel Y, Guedj D, Mukamel M, Harel L, Molad Y, Weinberger A (1999) Childhood Behcet’s disease: clinical features and comparison with adult-onset disease. Rheumatology 38(5):457–462
Sibley C, Yazici Y, Tascilar K, Khan N, Bata Y, Yazici H, Goldbach-Mansky R, Hatemi G (2014) Behcet syndrome manifestations and activity in the United States versus Turkey—a cross-sectional cohort comparison. J Rheumatol 41(7):1379–1384. https://doi.org/10.3899/jrheum.131227
Kobayashi T, Kishimoto M, Swearingen CJ, Filopoulos MT, Ohara Y, Tokuda Y, Oshikawa H, Yoshida K, Utsunomiya M, Kimura M, Okada M, Matsui K, Yazici Y (2013) Differences in clinical manifestations, treatment, and concordance rates with two major sets of criteria for Behcet’s syndrome for patients in the US and Japan: data from a large, three-center cohort study. Mod Rheumatol 23(3):547–553. https://doi.org/10.1007/s10165-012-0696-8
Moosmann T, Veraar C, Brunner J, Fraedrich G, Frech A, Horninger W, Ratzinger G, Streif W, Teuchner B, Willeit J, Zlamy M, De Zordo T, Schirmer M (2018) Differential clinical presentation of Adamantiades-Behcet’s disease in non-endemic and endemic areas: retrospective data from a Middle-European cohort study. Int J Rheum Dis. https://doi.org/10.1111/1756-185X.13306
Davatchi F, Shahram F, Chams-Davatchi C, Shams H, Nadji A, Akhlaghi M, Faezi T, Ghodsi Z, Faridar A, Ashofteh F, Sadeghi Abdollahi B (2010) Behcet’s disease: from East to West. Clin Rheumatol 29(8):823–833. https://doi.org/10.1007/s10067-010-1430-6
Batu ED, Sonmez HE, Sozeri B, Butbul Aviel Y, Bilginer Y, Ozen S (2017) The performance of different classification criteria in paediatric Behcet’s disease. Clin Exp Rheumatol 35(6):119–123
Assar S, Sadeghi B, Davatchi F, Ghodsi SZ, Nadji A, Shahram F, Ashofte F, Larimi SR, Sadeghi M (2017) The association of pathergy reaction and active clinical presentations of Behcet’s disease. Reumatologia 55(2):79–83. https://doi.org/10.5114/reum.2017.67602
Davatchi F, Sadeghi Abdollahi B, Chams-Davatchi C, Shahram F, Ghodsi Z, Nadji A, Akhlaghi M, Faezi T, Shams H, Larimi R, Ashofteh F (2013) Impact of the positive pathergy test on the performance of classification/diagnosis criteria for Behcet’s disease. Mod Rheumatol 23(1):125–132. https://doi.org/10.1007/s10165-012-0626-9
Faezi ST, Paragomi P, Shahram F, Shams H, Shams-Davatchi C, Ghodsi Z, Nadji A, Akhlaghi M, Davatchi F (2014) Clinical features of Behcet’s disease in patients without oral aphthosis. Mod Rheumatol 24(4):637–639. https://doi.org/10.3109/14397595.2013.844400
Mat MC, Goksugur N, Engin B, Yurdakul S, Yazici H (2006) The frequency of scarring after genital ulcers in Behcet’s syndrome: a prospective study. Int J Dermatology 45(5):554–556. https://doi.org/10.1111/j.1365-4632.2006.02859.x
Author information
Authors and Affiliations
Contributions
BED designed the structure of the article, drafted and critically revised the text, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Ezgi Deniz Batu declares that she has no conflict of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by the author.
Rights and permissions
About this article

Cite this article
Batu, E.D. Diagnostic/classification criteria in pediatric Behçet’s disease. Rheumatol Int 39, 37–46 (2019). https://doi.org/10.1007/s00296-018-4208-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-018-4208-9