
Abstract
We aimed to investigate whether the PTPRC rs10919563 A/G and Fc gamma receptor 2A (FCGR2A) R131H polymorphisms can predict the response to anti-TNF therapy in rheumatoid arthritis (RA) patients. We conducted a meta-analysis of studies on the association between the PTPRC rs10919563 A/G or the FCGR2A R131H polymorphism and responsiveness to anti-TNF therapy in RA patients. Eighteen studies (twelve on PTPRC and six on FCGR2A) from eight articles involving 3058 patients were considered in this meta-analysis. The meta-analysis showed a significant association between the PTPRC rs10919563 A allele and response to TNF-α blockers in RA. The OR of the PTPRC A allele was significantly lower in responders (OR = 0.584, 95 % CI = 0.409–0.835, P = 0.003). Meta-analysis revealed no association between the FCGR2A HH + HR genotype and responsiveness to TNF blockers in all study subjects (OR = 0.762, 95 % CI = 0.543–1.068, P = 0.115). However, stratification by TNF inhibitor type showed that the FCGR2A HH + HR genotype was associated with responsiveness to adalimumab (OR = 0.591, 95 % CI = 0.369–0.947, P = 0.029), but not infliximab and etanercept (OR = 0.929, 95 % CI = 0.354–2.440, P = 0.881; OR = 0.804, 95 % CI = 0.293–2.207, P = 0.673). The PTPRC rs10919563 A allele shows a poor response to anti-TNF therapy, and the FCGR2A HH + HR genotype shows a poor response to adalimumab for RA. Genotyping for these polymorphisms may be useful for predicting the response to TNF-α blockers with respect to personalized medicine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease that predominantly affects the synovial joints; up to 1 % of the world’s population has RA [1, 2]. Tumor necrosis factor-α (TNF-α) is a potent pro-inflammatory cytokine that plays an important role in inflammatory and immune responses, including those involved in RA [3]. Evidence regarding the central role played by TNF in the pathogenesis of RA has led to the introduction of anti-TNF therapy. TNF antagonists are among the most effective therapies for RA, but not all patients respond to treatment [4]. The reasons for this lack of response are unclear, but have generated considerable research interest because the prediction of response to TNF inhibitors would be highly valuable in preventing unnecessary biological therapy and reducing treatment costs, representing a significant advance in patient care.
The protein tyrosine phosphatase receptor type C (PTPRC, CD45) gene has been associated with RA susceptibility [5]. PTPRC is mapped at RA susceptibility loci, chromosome 1q31 proximal to genes encoding proteins involved in TNF signaling [5]. The PTPRC encoded by this gene is a member of the protein tyrosine phosphatase (PTP) family, which include signaling molecules that regulate a variety of cellular processes, including cell growth, differentiation, mitosis, and oncogenic transformation [6]. The PTPRC rs10919563 A/G polymorphism has been reported to be associated with response to TNF-α blockers in RA [7].
The Fc gamma receptors (FCGRs) play an important role in the recognition of immune complexes (ICs) [8]. FCGR2A is expressed on mononuclear phagocytes, neutrophils, and platelets, and the FCGR2A R131H (rs1801274) polymorphism is one of the most commonly studied FCGR polymorphisms in RA, as these allelic variations exhibit biological functions that differ among other FCGR genotypes [9]. Adalimumab, infliximab, and etanercept are the most commonly used TNF inhibitors in RA. These inhibitors have an IgG1 Fc portion that can bind to FCGRs [10, 11]. Thus, the genetic variants that affect the activity of FCGR2A depending on the different TNF blockers may influence the efficacy of TNF-α blockers [12].
Given the crucial roles played by the PTPRC and the FCGR2A polymorphisms in the pathogenesis of RA, many studies have examined the potential contributions of the PTPRC rs10919563 A/G and the FCGR2A R131H polymorphisms to the responsiveness to TNF antagonists in RA [11, 13–19]. However, these studies have often produced contradictory results. The disparities can be explained by small sample sizes, low statistical power, and clinical heterogeneity. A meta-analysis has been used to overcome the limitations of individual studies and resolve inconsistencies [20–22]. Using this approach, we investigated whether the PTPRC rs10919563 A/G and the FCGR2A R131H polymorphisms are associated with responsiveness to anti-TNF-α therapy in patients with RA.
Methods
Identification of eligible studies and data extraction
We considered all studies that examined the association between PTPRC rs10919563 A/G and/or FCGR2A R131H polymorphisms and responsiveness to anti-TNF-α therapy in patients with RA. A literature search was conducted using PubMed, EMBASE, and Cochrane Library. The search strategy included the following keywords and terms variably combined: “PTPRC,” “FCGR2A,” “polymorphism,” “TNF blocker,” “etanercept,” “infliximab,” “adalimumab,” and “rheumatoid arthritis.” Furthermore, the references of retrieved articles were manually reviewed to identify additional articles not found by electronic database searches. Language was not restricted. A study was included in the analysis if: (1) it was published before January 2016; (2) it presented original data (ensuring independence among studies); and (3) it provided sufficient data to calculate odds ratios (ORs). The exclusion criteria were as follows: the study (1) included duplicate data; (2) did not contain extractable data; and (3) had data on other polymorphisms. The following information was sought from each article: author identification, year of publication, country where the study was conducted, TNF-α blockers used, length of follow-up period, responsiveness criteria, and genotypes or alleles of the PTPRC rs10919563 A/G and FCGR2A R131H polymorphisms in responders and non-responders.
Evaluation of statistical associations
We calculated the overall estimate of the contrasts between A and G (allelic effect), AA versus AG + GG (recessive), AA + AG versus GG (dominant), and AA versus GG (homozygote contrast) models of the PTPRC rs10919563 A/G polymorphism with regard to responsiveness to TNF-α blockers. For the FCGR2A R131H polymorphism, the overall estimate of risk of H allele carriers (HH + HR) compared to the RR genotype for responsiveness to TNF-α blockers was calculated. Point estimates of risk, ORs, and 95 % confidence intervals (CI) were calculated for each included study. The Cochran’s Q test was used to assess within- and between-study variation and heterogeneity and test the null hypothesis that all studies evaluated the same effect. The effect of heterogeneity was quantified using the I 2 statistic, which ranges from 0 to 100 % and provides an estimate of the amount of total variability in point estimates attributable to heterogeneity rather than to chance [23]. I 2 values of 25, 50, and 75 % were designated as low, moderate, and high estimates, respectively. The fixed effects model assumes that a genetic factor has a similar effect on responders across all included studies and that observed variation among the studies is caused by chance alone [24]. Conversely, the random effects model assumes that different studies have substantial diversity and assesses both within-study sampling errors and between-study variance [25]. When the study groups are homogeneous, the two models are similar. In contrast, when the groups are heterogeneous, the random effects model usually provides wider CIs than the fixed effects model. The random effects model is best used when significant between-study heterogeneity is present [25]. The choice of meta-analysis model depends on the presence or absence of heterogeneity. In the absence of heterogeneity, a fixed effects model was used for meta-analysis. When a significant Q-value (P < 0.10) is calculated, indicating the existence of heterogeneity in the studies, a random effects model was utilized for meta-analysis. We scored the quality of each included study based on the Newcastle–Ottawa Scale (NOS) (25). A study was evaluated on three domains as follows: the selection of study groups (0–4 points), comparability of groups (0–2 points), and ascertainment of exposure (0–3 points). The highest score is 9, and a score in the range of 6–9 is considered to be of high methodological quality. Statistical analyses were performed using the Comprehensive Meta-Analysis program (Biostat, Englewood, NJ, USA). A P value <0.05 was considered statistically significant.
Evaluation of sensitivity test and publication bias
A sensitivity analysis was performed to assess the influence of each individual study on the pooled OR by omitting each study individually and to determine whether results from this meta-analysis were statistically robust. Funnel plots are used to detect publication bias, but require studies with different sizes that involve subjective judgments. Thus, we evaluated publication bias using Egger’s linear regression test [26], which measures funnel plot asymmetry on a natural logarithm scale of ORs.
Results
Studies included in the meta-analysis
Fifty-three studies were identified by electronic and manual searches, and 13 were selected for full-text review based on the title and abstract details. Five of these 13 studies were excluded because they did not include RA patients, had no data, or reported other polymorphisms. Thus, eight studies, consisting of 3058 patients with RA, met all the inclusion criteria and were considered in our meta-analysis [11, 13–19] (Fig. 1). One of the eligible studies contained data on nine different groups [16], and another study had data on three groups [11]; therefore, these groups were treated independently. Thus, 18 separate comparison groups were considered in the meta-analysis, and the sample sizes ranged from 43 to 689 patients. Twelve studies considered the PTPRC rs10919563 A/G polymorphism, and six studies investigated the FCGR2A polymorphism. The frequency of non-responders ranged from 17.6 to 46.4 % (mean 34.7 %), and follow-up periods ranged from 3 to 12 months. Quality assessment scores of the included studies ranged from 6 to 8, indicating high study quality (Table 1). Selected characteristics of these 18 studies are summarized in Table 1.

Flowchart used in study selection
Association of the PTPRC rs10919563 A/G polymorphism and responsiveness to TNF-α blockers
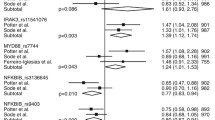
The meta-analysis showed a significant association between the PTPRC rs10919563 A allele and response to TNF-α blockers in RA (Table 2; Fig. 2). The OR of the A allele was significantly lower in responders (OR = 0.584, 95 % CI = 0.409–0.835, P = 0.003) (Table 2). However, the meta-analysis did not demonstrate an association between the PTPRC rs10919563 A/G polymorphism and response to TNF-α blockers under recessive, dominant, and homozygous contrast models (Table 2).

Odds ratios and 95 % confidence intervals of individual studies and pooled data for the associations between the PTPRC rs10919563 A allele and responsiveness to TNF blockers in patients with rheumatoid arthritis
Association of FCGR2A R131H polymorphism and responsiveness to TNF-α blockers
The meta-analysis revealed no association between the FCGR2A HH + HR genotype and responsiveness to TNF-α blockers in all study subjects (OR = 0.762, 95 % CI = 0.543–1.068, P = 0.115) (Table 3). However, stratification by TNF inhibitor type showed that the FCGR2A HH + HR genotype was associated with responsiveness to adalimumab (OR = 0.591, 95 % CI = 0.369–0.947, P = 0.029), but not infliximab and etanercept (OR = 0.929, 95 % CI = 0.354–2.440, P = 0.881; OR = 0.804, 95 % CI = 0.293–2.207, P = 0.673) (Table 3; Fig. 2). Stratification by follow-up time also failed to find a difference in the associations between the FCGR2A HH + HR genotype and responsiveness to TNF blockers in patients with short-term (<6 months) or long-term follow-up periods (Table 3).
Heterogeneity and publication bias
Between-study heterogeneity was found during the meta-analysis of the PTPRC rs10919563 A/G polymorphism (Table 2). I 2 values in the meta-analysis under allele contrast and dominant model were from moderate to high estimates, which means that the dispersion varies relatively widely among studies. However, there was no heterogeneity in recessive model and homozygous contrast. Sensitivity analysis showed that no individual study significantly affected the pooled OR, indicating statistically robust results from this meta-analysis. There was no heterogeneity in the meta-analyses of the FCGR2A R131H polymorphism in overall and adalimumab groups (Table 3). Funnel plots to detect publication bias were difficult to interpret because of the small number of studies in the meta-analysis. Egger’s regression test showed no evidence of publication bias in the meta-analysis (Egger’s regression test P values >0.1) (Fig. 3).

Odds ratios and 95 % confidence intervals of individual studies and pooled data for the associations between the FCGR2A R131H HH + HR genotype (B) and responsiveness to TNF blockers in patients with rheumatoid arthritis
Discussion
In the present study, we performed a meta-analysis of published data to look for differences in the PTPRC rs10919563 A/G or the FCGR2A R131H polymorphism between responders and non-responders to TNF blockers in RA. The meta-analysis revealed that patients with the PTPRC rs10919563 A allele had a poor responsiveness to TNF blockers compared to that in patients with the PTPRC G allele, and patients carrying the FCGR2A H allele had a poor response to adalimumab therapy compared to those with the FCGR2A RR genotype in RA.
This association of the PTPRC rs10919563 A/G or the FCGR2A R131H polymorphism with the responsiveness to anti-TNF-α therapy may be explained by various probable mechanisms. First, PTPRC, a receptor-like PTP expressed on immune-related cells, plays a critical role in TNF signaling by regulating T and B cell antigen receptor signaling, thus providing important regulation of signaling thresholds in immune-related cells [27, 28]. The PTPRC rs10919563 A/G polymorphism resides in intron 3, and intronic polymorphisms may have a functional role by influencing levels of gene expression [29]. Second, the PTPRC and FCGR2A genes, which map to the RA susceptibility loci of 1q31 and 1q21-23, respectively, have been identified as candidate genes [5, 30]. RA risk alleles may contribute to the responsiveness to anti-TNF therapy in RA [31]. Third, the FCGR2A R131H polymorphism also has functional significance. The FCGR2A R131H polymorphism results in either a histidine (H) or arginine (R) at position 131 within the second Ig-like domain of the receptor [32]. This affects the affinity of the receptor for IgG, with the H isoform having a higher affinity than the R isoform [33–35]. Neutrophils with the FCGR2A HH131 genotype efficiently bind to IgG2 with a threefold higher phagocytosis rate and have a sevenfold higher bactericidal activity than neutrophils with the RR131 genotype [33–35]. Therefore, the high-affinity H allele resulted in a higher clearance of TNF-α blockers from the circulation and thus decreased the response to treatment.
Our meta-analysis revealed that patients with the FCGR2A HH + HR genotype showed a poor response to adalimumab, but the association was not found in infliximab and etanercept therapy. The reason for this discrepancy is unclear, but it may be partly explained by low statistical power due to the small number of studies, because meta-analysis results on the association of the FCGR2A HH + HR genotype with responsiveness to three TNF blockers showed a same directionality. However, the sample size of study with infliximab, but not adalimumab, was the largest in the three TNF blockers studies. Another factors related to efficacy including the frequency or dosage of concomitant MTX, DAS28 level, or presence of rheumatoid factor might contribute to the discrepancy.
Some limitations in the present study warrant consideration. First, heterogeneity and publication bias may have distorted our meta-analysis because of the small number of studies included, especially in subgroup analyses. We could not rule out this possibility based on the small number of studies included in this meta-analysis. Second, confounders may also have affected the meta-analysis, as we could not adjust for clinical variables such as sex, disease severity, and disease duration, which may have influenced the response to anti-TNF-α therapy. Third, this meta-analysis included data only from European patients; therefore, these results are applicable only to this group. The relative importance of FCGR polymorphisms in disease susceptibility as well as in drug response may depend on ethnicity, as the allele frequency of the polymorphisms may differ between ethnic populations.
In conclusion, the present study shows that the PTPRC rs10919563 A/G polymorphism is associated with a response to anti-TNF therapy in RA, indicating that a patient with the PTPRC A allele has the potential to show a poor response to TNF-α blockers. In addition, the FCGR2A R131H polymorphism was associated with a poor response to adalimumab in RA, suggesting that individuals who carry the FCGR2A H allele are more likely to be less responsive to adalimumab therapy than those with the G allele in RA. The potential application of these results in a clinical setting is unclear because this meta-analysis focused on a limited number of studies and revealed between-study heterogeneity. Thus, further studies are needed to determine the effectiveness of these polymorphisms in predicting the efficacy of TNF blockers in patients with RA.
References
Harris ED Jr (1990) Rheumatoid arthritis. Pathophysiology and implications for therapy. N Engl J Med 322(18):1277–1289
Choi SJ, Rho YH, Ji JD, Song GG, Lee YH (2006) Genome scan meta-analysis of rheumatoid arthritis. Rheumatology (Oxford) 45(2):166–170
Brennan FM, Maini RN, Feldmann M (1992) TNF alpha–a pivotal role in rheumatoid arthritis? Br J Rheumatol 31(5):293–298
Salliot C, Finckh A, Katchamart W, Lu Y, Sun Y, Bombardier C et al (2011) Indirect comparisons of the efficacy of biological antirheumatic agents in rheumatoid arthritis in patients with an inadequate response to conventional disease-modifying antirheumatic drugs or to an anti-tumour necrosis factor agent: a meta-analysis. Ann Rheum Dis 70(2):266–271
Raychaudhuri S, Thomson BP, Remmers EF, Eyre S, Hinks A, Guiducci C et al (2009) Genetic variants at CD28, PRDM1 and CD2/CD58 are associated with rheumatoid arthritis risk. Nat Genet 41(12):1313–1318
Tonks NK (2006) Protein tyrosine phosphatases: from genes, to function, to disease. Nat Rev Mol Cell Biol 7(11):833–846
Cui J, Saevarsdottir S, Thomson B, Padyukov L, van der Helm-van Mil AH, Nititham J et al (2010) Rheumatoid arthritis risk allele PTPRC is also associated with response to anti-tumor necrosis factor alpha therapy. Arthritis Rheum Arthritis Care Res 62(7):1849–1861
Ravetch JV, Bolland S (2001) IgG Fc receptors. Annu Rev Immunol 19:275–290
Chai L, Song YQ, Leung WK (2012) Genetic polymorphism studies in periodontitis and Fcgamma receptors. J Periodontal Res 47(3):273–285
Kaymakcalan Z, Sakorafas P, Bose S, Scesney S, Xiong L, Hanzatian DK et al (2009) Comparisons of affinities, avidities, and complement activation of adalimumab, infliximab, and etanercept in binding to soluble and membrane tumor necrosis factor. Clin Immunol 131(2):308–316
Avila-Pedretti G, Tornero J, Fernández-Nebro A, Blanco F, González-Alvaro I, Cañete JD et al (2015) Variation at FCGR2A and functionally related genes is associated with the response to anti-TNF therapy in rheumatoid arthritis. PLoS One 10(4):e0122088
Arora T, Padaki R, Liu L, Hamburger AE, Ellison AR, Stevens SR et al (2009) Differences in binding and effector functions between classes of TNF antagonists. Cytokine 45(2):124–131
Ferreiro-Iglesias A, Montes A, Perez-Pampin E, Canete JD, Raya E, Magro-Checa C, et al (2016) Replication of PTPRC as genetic biomarker of response to TNF inhibitors in patients with rheumatoid arthritis. Pharmacogenomics J 16(2):137–140
Zervou MI, Myrthianou E, Flouri I, Plant D, Chlouverakis G, Castro-Giner F et al (2013) Lack of association of variants previously associated with anti-TNF medication response in rheumatoid arthritis patients: results from a homogeneous Greek population. PLoS One 8(9):e74375
Plant D, Prajapati R, Hyrich KL, Morgan AW, Wilson AG, Isaacs JD et al (2012) Replication of association of the PTPRC gene with response to anti-tumor necrosis factor therapy in a large UK cohort. Arthritis Rheum 64(3):665–670
Cui J, Saevarsdottir S, Thomson B, Padyukov L, van der Helm-van Mil AH, Nititham J et al (2010) Rheumatoid arthritis risk allele PTPRC is also associated with response to anti-tumor necrosis factor alpha therapy. Arthritis Rheum 62(7):1849–1861
Davila-Fajardo CL, van der Straaten T, Baak-Pablo R, Medarde Caballero C, Cabeza Barrera J, Huizinga TW et al (2015) FcGR genetic polymorphisms and the response to adalimumab in patients with rheumatoid arthritis. Pharmacogenomics 16(4):373–381
Montes A, Perez-Pampin E, Narvaez J, Canete JD, Navarro-Sarabia F, Moreira V et al (2014) Association of FCGR2A with the response to infliximab treatment of patients with rheumatoid arthritis. Pharmacogenet Genomics 24(5):238–245
Canete JD, Suarez B, Hernandez MV, Sanmarti R, Rego I, Celis R et al (2009) Influence of variants of Fc gamma receptors IIA and IIIA on the American College of rheumatology and european league against rheumatism responses to anti-tumour necrosis factor alpha therapy in rheumatoid arthritis. Ann Rheum Dis 68(10):1547–1552
Lee YH, Rho YH, Choi SJ, Ji JD, Song GG (2007) PADI4 polymorphisms and rheumatoid arthritis susceptibility: a meta-analysis. Rheumatol Int 27(9):827–833
Lee YH, Bae SC, Choi SJ, Ji JD, Song GG (2011) Associations between vitamin D receptor polymorphisms and susceptibility to rheumatoid arthritis and systemic lupus erythematosus: a meta-analysis. Mol Biol Rep 38(6):3643–3651
Lee YH, Woo JH, Choi SJ, Ji JD, Song GG (2010) Associations between osteoprotegerin polymorphisms and bone mineral density: a meta-analysis. Mol Biol Rep 37(1):227–234
Higgins JP, Thompson SG (2002) Quantifying heterogeneity in a meta-analysis. Stat Med 21(11):1539–1558
Egger M, Smith GD, Phillips AN (1997) Meta-analysis: principles and procedures. BMJ 315(7121):1533–1537
DerSimonian R, Laird N (1986) Meta-analysis in clinical trials. Control Clin Trials 7(3):177–188
Egger M, Davey Smith G, Schneider M, Minder C (1997) Bias in meta-analysis detected by a simple, graphical test. BMJ 315(7109):629–634
Hermiston ML, Xu Z, Weiss A (2003) CD45: a critical regulator of signaling thresholds in immune cells. Annu Rev Immunol 21:107–137
Stanford SM, Rapini N, Bottini N (2012) Regulation of TCR signalling by tyrosine phosphatases: from immune homeostasis to autoimmunity. Immunology 137(1):1–19
Cooper DN (2010) Functional intronic polymorphisms: buried treasure awaiting discovery within our genes. Hum Genomics 4(5):284–288
Choi SJ, Rho YH, Ji JD, Song GG, Lee YH (2006) Genome scan meta-analysis of rheumatoid arthritis. Rheumatology 45(2):166–170
Tak PP (2012) A personalized medicine approach to biologic treatment of rheumatoid arthritis: a preliminary treatment algorithm. Rheumatology 51(4):600–609
Su K, Wu J, Edberg JC, McKenzie SE, Kimberly RP (2002) Genomic organization of classical human low-affinity Fcgamma receptor genes. Genes Immun 3(Suppl 1):S51–S56
Sanders LA, Feldman RG, Voorhorst-Ogink MM, de Haas M, Rijkers GT, Capel PJ et al (1995) Human immunoglobulin G (IgG) Fc receptor IIA (CD32) polymorphism and IgG2-mediated bacterial phagocytosis by neutrophils. Infect Immun 63(1):73–81
Nimmerjahn F, Ravetch JV (2008) Fcγ receptors as regulators of immune responses. Nat Rev Immunol 8(1):34–47
Catrina AI, Trollmo C, af Klint E, Engstrom M, Lampa J, Hermansson Y et al (2005) Evidence that anti-tumor necrosis factor therapy with both etanercept and infliximab induces apoptosis in macrophages, but not lymphocytes, in rheumatoid arthritis joints: extended report. Arthritis Rheum 52(1):61–72
Acknowledgments
This study was supported in part by a grant of the Korea Healthcare Technology R&D Project, Ministry for Health and Welfare, Republic of Korea (HI13C2124).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Young Ho Lee declares no conflict of interest. Sang-Cheol Bae declares no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Rights and permissions
About this article

Cite this article
Lee, Y.H., Bae, SC. Associations between PTPRC rs10919563 A/G and FCGR2A R131H polymorphisms and responsiveness to TNF blockers in rheumatoid arthritis: a meta-analysis. Rheumatol Int 36, 837–844 (2016). https://doi.org/10.1007/s00296-016-3476-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-016-3476-5