
Abstract
Eukaryotic DNA replication is accompanied by the disassembly and reassembly of nucleosomes and the transmission of epigenetic marks to the newly assembled chromatids. Several histone chaperones, including CAF-1 and Asf1p, are central to these processes. On the other hand, replication forks pause at numerous positions throughout the genome, but it is not known if and how this pausing affects the reassembly and maintenance of chromatin structures. Here, we applied drug-free gene silencing assays to analyze the genetic interactions between CAC1, ASF1, and two genes that regulate the stability of the paused replisome (TOF1) and the resumption of elongation (RRM3). Our results show that TOF1 and RRM3 differentially interact with CAF-1 and ASF1 and that the deletions of TOF1 and RRM3 lead to reduced silencing and increased frequency of epigenetic conversions at three loci in the genome of S. cerevisiae. Our study adds details to the known activities of CAF-1 and Asf1p and suggests that the pausing of the replication fork can lead to epigenetic instability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gene silencing in S. cerevisiae has served as a paradigm for complex chromatin-mediated phenomena in other eukaryotes. In this organism, the silencing of genes at the mating type loci HMRa and HMLα, the sub-telomeric regions of the chromosomes, and the rRNA-encoding DNA (the rDNA) have received significant attention (Sauty et al. 2021; Shaban et al. 2021). At these positions, the Histone Deacetylase (HDAC) Sir2p and the associated Silent Information Regulator (Sir) proteins initiate and maintain a cascade of spreading of histone deacetylation along the adjacent nucleosomes (Rusche et al. 2003). This activity leads to a meta-stable alternating between active and silent states of reporter genes inserted in the sub-telomeres or rRNA gene loci (Rusche et al. 2003). The mating type loci are robustly repressed and constitutively silent but can show alternating active/silent states upon compromised silencing (Rusche et al. 2003; Yankulov 2013). A similar meta-stable phenotype is observed at the FLO genes loci; however, the silencing at these positions is independent of the SIR genes (Sauty et al. 2021; Shaban et al. 2021).
The mechanisms of SIR-mediated gene silencing are well characterized (Gartenberg and Smith 2016). In comparison, the mechanisms of maintenance of the epigenetic state through multiple rounds of cell divisions and passages of the replication forks are insufficiently addressed (Rowlands et al. 2017). The current models for histone turnover during DNA replication and the preservation of epigenetic state have been recently summarized in (Stewart-Morgan et al. 2020; Shaban et al. 2021). Briefly, as the replication fork advances, the nucleosomes are disassembled and the “old” histones are ferried behind the fork through the activity of the Asf1p and FACT histone chaperones. Behind the fork another histone chaperone, the Chromatin Assembly Factor 1 (CAF-1), is assembling H3/H4 tetramers. Recently, it has been shown that the MCM helicase, and the Dbp3–Dbp4 subunits of DNA polymerase ε and Ctf4p are involved in the symmetric distribution of H3/H4 tetramers (Gan et al. 2018; Petryk et al. 2018; Yu et al. 2018), thus leading to the suggestion that the CAF-1 function is restricted to filling-in the gaps by assembling H3/H4 from “new” dimers delivered from the cytoplasm (Ahmad and Henikoff 2018; Groth et al. 2007; Almouzni and Cedar 2016). However, earlier studies have suggested that CAF-1 could have additional functions at certain loci where the “old” H3/H4 tetramers can be lost (Almouzni and Cedar 2016; Rowlands et al. 2017). After the reassembly of nucleosomes behind the fork, the epigenetic marks on the old histones are copied onto the new ones by a largely unknown mechanism (Ahmad and Henikoff 2018).
It is known that replication forks slow down and/or pause at numerous positions occupied by proteins that are tightly bound to DNA or at active gene promoters (Ivessa et al. 2003; Makovets et al. 2004; Azvolinsky et al. 2006; Shyian and Shore 2021). Frequent fork pausing has been detected in the rRNA gene loci and in the sub-telomeres, especially in the absence of Rrm3p, a DNA helicase necessary for the release of the paused forks in vivo but not for efficient fork progression in experiments conducted with purified/recombinant proteins in vitro (Azvolinsky et al. 2006; Deegan et al. 2019). Analyses of fork stalling at natural or engineered protein barriers have revealed that mammalian homologues of RRM3 (RTEL1, DDX11, FANCJ, and DHX36) and other helicases or scaffold proteins (BLM and FANCM) operate to restart/repair the stalled replisomes (Shyian and Shore 2021; Scully et al. 2021). Forks also stall at sites of DNA damage and possibly at sites of stable secondary DNA structures (Shyian and Shore 2021). However, the role of RRM3 and its homologues under these conditions is not clear.
The Fork Protection Complex (FPC, built-up of Mrc1p/Csm3p/Tof1p) is known to stabilize the paused forks and ensure the timely resumption of elongation. The FPC is also crucial for processive elongation in vitro (Yeeles et al. 2017). Additional functions of Tof1p include the recruiting of FACT to the fork (Safaric et al. 2022) and linking FPC to the function of Topoisomerase I (TOP1). More specifically, the C-terminus of Tof1p is required for the recruitment of Top1p to proteinaceous fork barriers, but not for the activation of DNA replication checkpoints (Shyian et al. 2020). Prior genetic studies have demonstrated that the effects of TOF1 deletion at the rRNA gene arrays are counteracted by deletion of RRM3 (Mohanty et al. 2006; Bastia et al. 2016). However, a recent study added that the functions of RRM3 and TOF1 can be separated in vivo, suggesting that these factors act independently of each other (Shyian et al. 2020). How the activities of RRM3 and TOF1 and fork pausing affect histone turnover and the maintenance of chromatin state remains unclear.
We have recently shown that the deletion of RRM3 exacerbated the silencing defects observed at the telomeres and at the FLO11 loci upon the deletion of CAC1 (the gene that encodes the largest subunit of CAF-1) (Wyse et al. 2016; Rowlands et al. 2017). In this study, we asked how TOF1 and RRM3 genetically interact with the H3/H4 histone chaperones involved in the replication-coupled reassembly of nucleosomes.
Materials and methods
Yeast strains
Strains with single gene deletions were obtained from Open Biosystems or were gifts from other labs (see S1 Table). Double deletion mutants of tof1 with cac1, rrm3, and asf1 were generated by direct knockout of TOF1 with PCR fragments or by routine mating/sporulation lab techniques. The tof1-726∆ and the tof1-830∆ derivatives were produced by PCR-mediated knockout of ASF1, CAC1, and RRM3 in the strains provided by (Westhorpe et al. 2020). All gene deletions were confirmed by PCR prior and after each genetic manipulation. JRY0803 and its derivatives were used in the CRASH assay as in (Janke et al. 2018). All strains were routinely maintained at 30 °C on Yeast Extract Peptone Dextrose YPD (1% yeast extract, 2% tryptone, and 2% glucose) media. Flow cytometry and fluorescent microscopy experiments were conducted with cells grown in Synthetic Complete (SC) media and SC drop-out media, as required.
Growth rates’ analysis
Strains were serially diluted in a 96-well plate and the OD600 of the exponentially growing cultures was measured every 2 h for a period of 24 h using infinite 200Pro plate reader with continuous shaking. The data were analyzed in the MS Excel®.
Methyl methane sulfonate (MMS) sensitivity
Cells from exponentially growing cultures were serially diluted in a 96-well tray plate and 5 µl aliquots were spotted on plates with different MMS concentrations [0.05%, 0.1% and 0.2% MMS (Sigma)]. Plates were incubated at 30 °C for 2 days and images were taken by regular photography.
Cell cycle analysis
Cells in early exponential phase were arrested by 15 μg/mL Nocodazole for three hours at 30 °C. Cells were washed twice and resuspended in YPD medium, and samples were collected at different time points after the release from the block. Cells were then fixed with 70% Ethanol and DNA was stained with Propidium Iodide as in (Jeffery et al. 2015). Cell cycle analysis was done using Sony SH800z flow cytometer and LESH00SZFCPL™ software.
HTB1-yEGFP-URA3-tel, FLO11-yEGFP, and CRASH reporters
An yEGFP reporter driven by the Histone H2B promoter (HTB1) (HTB1-yEGFP) from pFOM298 plasmid (Mano et al. 2013) was cloned into the pUCAIV plasmid (Gottschling et al. 1990) to produce the ADH4-HTB1-yEGFP-URA3-tel construct for insertion into the VIIL telomere. The HTB1-yEGFP reporter was inserted in both orientations relative to the telomere (Fig S1.A). It was found that the reverse orientation toward telomere produced higher proportions of GFP + cells and signals and all subsequent experiments were conducted with this construct. The FLO11-yEGFP reporter was produced by replacing the FLO11 ORF at its genome location with yEGFP-KanMX construct as described in (Rowlands et al. 2019b) (Fig S1.B). The strains for CRASH analyses contain an expression cassette for the CRE recombinase inserted in the HML locus as described in (Janke et al. 2018) (Fig S1.C). Genome manipulation in all strains was confirmed by PCR.
Analysis of yEGFP signals by fluorescent microscopy
Cell suspensions were grown to saturation in SC media. After vigorous vortexing to disperse cell clusters, images were acquired by non-confocal Leica DM 6000B microscope with bright field and fluorescent field at 40X and 100X immersion oil lenses. Alternatively, cells’ cultures were serially diluted in a 96-well plate and incubated at 25 °C overnight, and images were acquired by Diskovery™ Spinning Disk confocal microscope with 60X lens. All images were processed and analyzed by Volocity™ software as in (Rowlands et al. 2019b). Briefly, the pixel values of at least 25 ROIs (Region of Interest) with no cells were used to calculate the average background for each individual experiment. Next, pixel values in at least 25 ROI over whole single isolated cells with no visible signal in them were acquired and used as a postulated arbitrary threshold line. Finally, pixel values of ROI over all cells in three separate fields with 50–100 cells were measured. Distribution graphs of the raw intensity signals are shown in Supplemental Materials (Fig. S4).
The accuracy of the postulated threshold lines between experiments was evaluated by the prediction approaches of the statistical decision theory (Hastie et al. 2009). This algorithm showed that a threshold level of 160% of the average background identifies positive cells with 100% accuracy (Fig S2, S3). For negative cells, accuracy was greater than 90% (Fig S2, S3). We used this threshold in each individual experiment with all strains tested. At least three separate experiments were conducted with each strain. Average numbers and standard deviations were calculated in Excel®.
CRASH (cre-reported altered states of heterochromatin) assays
The CRASH assay was performed as described previously (Janke et al. 2018; Rowlands et al. 2019b). The strains for this assay contain an RFP/yEGFP switch reporter cassette that converts the expression of RFP to yEGFP upon the expression of CRE (Fig. S1.C). Briefly, cells were grown on the appropriate SC drop-out plates containing 300 μg/mL hygromycin to select for the initial 100% RFP positive state. A single colony from this plate was dispersed in SC medium and cells were plated on appropriate drop-out media based on their knockout markers at 50–100 colonies per plate. Colonies were imaged after 5–7 days growth using a Zeiss AxioZoom V16 microscope at 2X magnification equipped with a Hamamatsu Camera and Zen software. The de-repression events for each single and double mutant could be visualized by green segments that could be counted. Counting of green segments was done for 4–5 individual colonies from each strain except for some double mutants where the multiplicity of green segments precludes accurate counts. Statistical analyses were performed, and graphs were built using the MS Excel®.
Flow cytometry
Cells were grown until they reached an exponentially growing state of OD600 = 1.0, and cells were harvested and followed by sonication to be dispersed. They were suspended in Phosphate-Buffered Saline (PBS) prior to analysis by a Sony SH800z flow cytometer. The LESH00SZFCPL™ software was used to collect data. Isogenic strains with no yEGFP reporters were used to set up the gates for GFP-negative cells. Three independent experiments were conducted with each strain. Graphs were produced and analyzed by the MS Excel®.
Measurement of conversions rates
Strains were grown in SC medium and briefly sonicated to disperse cell aggregates. The cultures were then diluted to about 3–5 cell/ml and 200 µl aliquots were dispensed in three 96-well plates. The plates were incubated at 25 °C until a single cluster of cells could be seen at low magnification of the microscope. Under these conditions, most of these mini-cultures originate from a single cell and go through about 8–11 generations. Cells from single clusters were then dispersed and analyzed by flow cytometry to determine the percentage of GFP + cells in each colony. Data analysis and Graphs were made in the MS Excel®.
Results
Experimental strategy
It has been previously shown that the loss of gene silencing at the sub-telomeric and FLO11 loci in cac1∆ and asf1∆ strains is exacerbated by the deletion of RRM3 (Wyse et al. 2016; Rowlands et al. 2019b) and that the deletions of RRM3 or TOF1 have opposite effects on the pausing of replication forks at the rRNA gene array (Mohanty et al. 2006). It remains unclear if and how TOF1 is contributing to gene silencing. To address this question, we produced a series of double deletion mutants of CAC1, ASF1 along with RRM3 and TOF1. We attempted the production of cac1∆mrc1∆ and asf1∆mrc1∆ mutants, but these strains were not viable. In the viable strains, we inserted yEGFP reporters in two loci that display gene silencing and on/off “variegated” patterns of gene expression. At the FLO11 locus, the reporter is driven by the FLO11 promoter itself (Rowlands et al. 2019b). In the VIIL telomere, we inserted the URA3-HTB1-yEGFP1 construct in which yEGFP is driven by the strong Histone H2B promoter (HTB1) (Mano et al. 2013; Shaban et al. 2023). This construct faithfully recaptures most of the effects observed previously by URA3 reporters and precludes the use of the toxic 5-FOA (Mano et al. 2013; Shaban et al. 2023). It also provides strong yEGFP signals that can be evaluated by drug-free flow cytometry and/or fluorescent microscopy assays. Previous reports had indicated that the repression of the native HTB1 gene was dependent on the activity of ASF1 and other histone chaperones and the adjacent Yta7 chromatin boundary (Sutton et al. 2001; Zunder and Rine 2012). However, the HTB1-yEGFP construct does not contain the boundary and in our hands does not demonstrate sensitivity to the deletion of ASF1, while the deletion of ASF1 reduced silencing at the FLO11 and HML loci (see below).
At the mating HMLα locus, we measured the levels of silencing by a CRE expression cassette and the CRASH assay (Janke et al. 2018; Brothers and Rine 2019).
At the VIIL and HMLα loci, gene silencing is critically dependent on the recruitment and activity of Sir2p HDAC, while FLO11 silencing is independent of the SIR genes.
Characteristics of the double deletion mutants.
First, we checked some general characteristics of the produced strains. Growth rates were measured in liquid cultures in 96-well trays on a shaking platform supplied with a spectrophotometer. In agreement with the previous studies, the cac1∆, rrm3∆, tof1∆, and asf1∆ strains showed little loss in growth rates as compared to the isogenic BY4742 strain (Park and Sternglanz 1999; Jeffery et al. 2013; Wyse et al. 2016) (Fig. 1A). The double mutants tof1Δcac1Δ and tof1Δrrm3Δ also did not show any major growth defects. However, a decrease in growth rate was detected in the tof1Δasf1Δ double mutant (Fig. 1A). Analyses of cell cycle progression after arrest with Nocodazole revealed that the reduced growth rate of tof1Δasf1Δ cultures should be attributed to a very slow progression through G1/S phases (Fig. 1B). The tof1∆ cells showed minor cell cycle defects as compared to BY4742. Interestingly, asf1∆ cells were slow in exiting mitosis. At present, we have no good explanation for this effect of asf1∆.

Characteristics of the mutant strains. (A) Growth rates analysis. Cells from exponentially growing cultures were diluted in a 96-well plate and the OD600 was measured every 2 h for a period of 24 h. The graphs were made using the MS Excel®. (B) Cell Cycle analysis. Exponentially growing cells were arrested by Nocodazole and then released from the arrest, and samples were collected at the indicated time points and stained with Propidium Iodine. (C) MMS sensitivity. Cells from an exponentially growing cultures were serially diluted and aliquots were spotted on plates with different concentrations of MMS (0.005%, 0.01%, and 0.02%). One of two independent experiments is shown
All strains were also tested for sensitivity to DNA damage by exposure to 0.05%, 0.1%, and 0.2% MMS (Fig. 1C). MMS methylates guanine and adenine bases leading to mispairing, replication blocks, point mutations, and double-stranded DNA breaks (Beranek et al. 1983). We used a mrc1∆ strain as a control. Consistent with the previous studies, the deletions of CAC1, RRM3, and ASF1 marginally increased sensitivity to this mutagen (Tyler et al. 1999; Jeffery et al. 2013; Wyse et al. 2016). The deletions of TOF1 and MRC1 had a stronger effect relative to the cac1∆, asf1∆, and rrm3∆ strains. The double deletions tof1Δcac1Δ and tof1Δrrm3Δ did not display higher sensitivity as compared to the single mutants. However, the deletion of both TOF1 and ASF1 lead to an additive sensitivity to this type of DNA damage, thus reiterating a possible ASF1-TOF1 functional link. Similar increased sensitivity to DNA damage has been earlier observed in cac1Δrrm3Δ (Rowlands et al. 2019b).
We also noticed that the tof1Δasf1Δ strain formed small clusters of cells in liquid cultures (Fig. 2D). The clusters were smaller than the ones previously observed in cac1Δasf1Δ, rrm3Δasf1Δ, and cac1Δrrm3Δ strains (Rowlands et al. 2019b). This mild flocculation phenotype was only moderately enhanced in the presence of Nicotinamide (not shown). None of the other strains displayed flocculation.

Analyses of the expression of HTB1-yEGFP at the VIIL telomere. (A) Proportions of GFP+ cells (%) in various mutants as measured by flow cytometry. (B) Proportions of GFP+ cells (%) in various mutants as measured by fluorescent microscopy. Distribution graphs of the raw intensity signals are shown in Fig. S4, A. (C) Flow cytometry graphs of yEGFP+ cells in various mutants. Cells were analyzed by Sony SH800z flow cytometer and graphs were generated by The LESH00SZFCPL™ Software. Gating for GFP-negative cells was based on the identical analyses of isogenic strains without the HTB1-yEGFP reporter. (D) Representative microscopy images of various mutants. Cells were analyzed by Leica DM 6000B microscopy and signals were processed by the Volocity® software. (E) Proportions of GFP+ cells (%) in TOF1, tof1-762∆, and tof1-762∆ strains as measured by flow cytometry. (F) Flow Cytometry graphs of yEGFP+ cells in tof1 mutants. * represents statistically significant difference compared to the BY4742 strain. ** represents statistically significant difference between the two connected stains (independent T tests were used)
Analysis of HTB1-yEGFP expression at the VIIL telomere
Previous studies have pointed out that the deletion of CAC1 and, to less extent, the deletion of ASF1 and RRM3 lead to loss of silencing at the VIIL telomere as measured by a URA3 reporter and sensitivity to 5-FOA (Zhang et al. 2000; Jeffery et al. 2013; Wyse et al. 2016). We revisited these phenotypes by Flow Cytometry and fluorescent microscopy assays with the URA3-HTB1-yEGFP-tel construct (Fig. 2). We also tested the effects of deletion of TOF1 in BY4742, cac1∆, asf1∆, and rrm3∆ genetic backgrounds. In these assays, the increase of the percentage of GFP-positive cells indicates loss of silencing and the decrease indicates reversal of the loss.
In agreement with the previous studies, in both flow cytometry and microscopy experiments, the deletion of CAC1 was accompanied by an increase in the percentage of yEGFP+ cells (Fig. 2A, B). The deletion of ASF1 did not show statistically significant changes in the expression of the HTB1-yEGFP construct as compared to BY4742 (Fig. 2A, B). Surprisingly, and in disagreement with prior experiments with the URA3/5-FOA assays, in the rrm3∆ strain, the flow cytometry analyses indicated a statistically significant decrease in the proportion of yEGFP+ cells (Fig. 2A), while fluorescent microscopy detected no difference between the BY4742 and rrm3∆ cells (Fig. 2B). It is possible that these discrepancies are caused by the effect of 5-FOA on the pools of dNTPs (Rossmann et al. 2011; Takahashi et al. 2011) in conjunction with the deletion of RRM3. The deletion of TOF1 also decreased in the proportion of yEGFP+ cells as measured by flow cytometry but not by microscopy (Fig. 2A, B). Hence, a possible anti-silencing effect of the individual deletions of RRM3 or TOF1 at the VIIL telomere seems subtle and is not conclusively established by our yEGFP-based assays.
Next, we tested the silencing of the HTB1-yEGFP reporter in the double deletion mutants. The deletion of TOF1 in the cac1∆ strain increased silencing as demonstrated by the reduction in the proportion of yEGFP+ cells (Fig. 2A, B). Conversely, in the asf1∆ background the deletion of TOF1 increased the proportion of yEGFP + cells suggesting a decreased silencing of the HTB1-yEGFP reporter (Fig. 2A, B). These results demonstrate that the TOF1 and RRM3 genes, similar to their effect on paused replication forks (Mohanty et al. 2006), have opposite functions in the silencing at the VIIL telomere. In the rrm3∆ background, the deletion of TOF1 increased the proportion of yEGFP+ as measured by flow cytometry, but this result was not confirmed by the microscopy experiments.
In agreement with our prior studies, we observed a substantial loss of gene silencing at the VIIL telomere in the cac1Δrrm3∆, asf1Δcac∆, and asf1Δrrm3∆ mutants by both flow cytometry and fluorescent microscopy (Fig. 2A, B), but not to the extent detected by the URA3/5-FOA assay (Jeffery et al. 2013; Wyse et al. 2016; Rowlands et al. 2019b).
Earlier structure–function analyses of TOF1 had demonstrated that the deletion of its C-terminus beyond amino acid 762 (tof1-762∆) precluded its interaction with Csm3p, the pausing of the fork, and the TOF-mediated sensitivity to DNA damage (Westhorpe et al. 2020). The truncation of the protein at position 830 (tof1-830∆) displayed significantly milder phenotypes and did not preclude fork pausing (Westhorpe et al. 2020).
To get further insights in the mechanism of TOF1-mediated maintenance of gene silencing, we tested if these tof1 mutants alter the expression of HTB1-yEGFP (Fig. 2E). The truncation at position 830 (tof1-830∆) did not significantly change the expression of HTB1-yEGFP (Fig. 2E). In this genetic background, the deletions of CAC1 and ASF1 reduced silencing as demonstrated by the higher proportions of GFP + cells, while the deletion of RRM3 did not have a statistically significant effect (Fig. 2E). However, the truncation at position 762 (tof1-762∆) markedly reduced the silencing of the construct. In this strain, the deletions of ASF1 and RRM3 in this strain marginally increased the proportions of GFP + cells, while the deletion of CAC1 had a statistically significant effect (Fig. 2E). We conclude that the tof1-762∆ allele, which is known to reduce the pausing of replication forks (Westhorpe et al. 2020), also reduced the silencing of the HTB1-yEGFP construct. At the same time, the genetic interactions of the tof1-762 allele with ASF1, CAC1, and RRM3 did not precisely phenocopy the effects of these genes observed with the complete destruction of TOF1 (Fig. 2A).
Analyses of gene silencing at the FLO11 locus
The analyses of gene silencing at the FLO11 locus were performed using a yEGFP reporter driven by the FLO11 promoter (FLO11-yEGFP). Again, the increase of the percentage of GFP-positive cells indicates loss of silencing and the decrease indicates reversal of the loss. In the flow cytometry experiments, the single mutants cac1∆, asf1∆, rrm3∆, and tof1∆ revealed no statistically significant increase in the expression of yEGFP relative to BY4742 cells (Fig. 3A) and modest, but statistically significant increase in the proportions of GFP + cells in the microscopy experiments (Fig. 3B). Importantly, the deletion of TOF1 in any of the cac1∆, asf1∆, and rrm3∆ strains had no substantial effect on the silencing of FLO11-yEGFP. On the other hand, the combination of deletions of CAC1, ASF1, and RRM3 lead to a substantial increase in the number of GFP + cells. This result is in agreement with our earlier study (Rowlands et al. 2019b) in which FLO11-mRNA expression was substantially increased in bulk cultures of these double deletion mutants. In the current study, we reproduce this observation at a single cell resolution using a yEGFP reporter.

Analyses of the expression of FLO11-yEGFP. (A) Proportions of GFP+ cells (%) in various mutants as measured by flow cytometry. * represents statistically significant difference compared to the BY4742 strain. ** represents statistically significant difference between the two connected stains (independent T tests were used). (B) Proportions of GFP+ cells (%) in various mutants as measured by fluorescent microscopy. Distribution graphs of the raw intensity signals are shown in Fig. S4, B. (C) Flow Cytometry graphs of yEGFP+ in various mutants. Cells were analyzed by Sony SH800z flow cytometry and graphs were generated by LESH00SZFCPL™ Software. Gating for GFP-negative cells was based on the identical analyses of isogenic strains without the HTB1-yEGFP reporter. (D) Representative microscopy images of various mutants. Cells were analyzed by Leica DM 6000B microscopy and signals were processed by the Volocity® software
These results and the very modest flocculation phenotype in asf1∆tof1∆ cells point out that TOF1 could not play a major role at this silenced locus.
Analysis of gene silencing the HMLα locus
The silencing at the HMLα locus was performed by the CRASH assay as in (Janke et al. 2018). The assay utilizes a Cre recombinase inserted at HML and a separate RFP → yEGFP reporter cassette on chromosome V. Transient de-repression of HMLα would trigger the Cre-mediated removal of RFP and the expression of yEGFP. This irreversible effect is visualized as green segments in red colonies (Dodson and Rine 2015; Janke et al. 2018). In these assays, the increase in the number of green segments in a red colony indicates transient loss of silencing. The appearance of totally green colonies indicates loss of silencing beyond the quantitative scope of the assay. Previous studies have demonstrated that cac1∆ cells do not display mating defects or de-repression of yEGFP reporters inserted in HMLα (Jeffery et al. 2013). In contrast, studies with the CRASH assay in cac1∆ revealed numerous green segments (Janke et al. 2018; Rowlands et al. 2019b). We applied this high sensitivity assay in the mutants generated in this study. Average numbers of green segments in 4–5 colonies of each strain were calculated and plotted (Fig. 4A). Compared to BY4742, the rrm3∆, asf1∆, and tof1∆ colonies showed statistically significant increase in the number of green segments, with asf1∆ showing the least effect (Fig. 4A, B). The segments in cac1∆ cells were too numerous to count indicating a more profound silencing defect.

Analyses of gene silencing at HMLα by CRASH assay. (A) Cells were grown in the presence of Hygromycin to select for the RFP+ state of the RFP → yEGFP reporter, then streaked on SC drop-out or SC/Geniticin agar, as appropriate, and grown to produce visible colonies. Images of colonies were taken with Axiozoom microscope using green and red filters and merged in Zen Software. Green sectors in 3–5 colonies were counted and plotted. ** represents statistically significant difference between the two connected stains (independent T tests were used). (B) Images of sectored colonies in various mutants
TOF1 again showed differential effects on gene silencing depending on the genetic background. In the cac1∆ background, the deletion of TOF1 led to a lower countable number of the green segments thus indicating reversal of the loss of silencing (Fig. 4A, B). In contrast, the tof1∆-mediated silencing defects were exacerbated in the asf1∆ strain. In the rrm3∆ strain, the deletion of TOF1 also increased the number of green segments, but to a lesser extent as compared to asf1∆ (Fig. 4A, B). The cac1Δasf1Δ, cac1Δrrm3Δ, and asf1Δrrm3Δ double mutants displayed predominantly green colonies thus pointing to a profound defect in HMLα silencing. We conclude that both RRM3 and TOF1 contribute to the silencing of HMLα. Similarly to the situation at the VIIL telomere (Fig. 2), they can also reduce or increase the silencing deficiencies when combined with deletions of CAC1 and ASF1, respectively.
Conversion frequencies at the VIIL telomere
Positional variegation is characterized by infrequent Silent → Active and Active → Silent (S → A and A → S) conversions of the affected loci (Yankulov 2013). In an earlier study, we have used the URA3/5-FOA assay to show that the loss of CAC1 could lead to lower frequency of such epigenetic conversions (Jeffery et al. 2013). Here, we have tested the effect of our mutants on the frequency of conversions of the HTB1-yEGFP reporter at the VIIL telomere. We applied a technique that was identical to the one previously used to assess the frequency of conversions of a different yEGFP reporter inserted at the HMRa locus (Jeffery et al. 2013). Briefly, cells were serially diluted in 96-well plates and grown for 8–10 generations. Mini-cultures in wells with single clusters of cells were deemed to originate from a single cell. These mini-cultures were analyzed by flow cytometry and the percentage of GFP+ cells in each of them was plotted as a single bar in a plot of multiple bars (Fig. 5). The number of generations in each mini-culture was determined by the count of events in the flow cytometer and any mini-culture with less than eight or more than ten generations was excluded from further analyses. Between 30 and 50 mini-cultures were analyzed for each strain.

Measurements of conversion rates of at the VIIL telomere. Mini-cultures originating from a single cell were grown for 8–10 generations and analyzed by flow cytometry as in Fig. 2A. Each bar represents the %GFP+ cells in an individual colony. The last bar on the right represents the %GFP+ cells in the seeding culture. (A) A simulation of a strain with 30% GFP + cells in the seeding culture and 9% A → S and 5% S → A conversion rates per generation. (B) A simulation of a strain with 60% GFP + cells in the seeding culture and 5% A → S and 3% S → A conversion rates. (C–H) Conversion rates in the strains indicated on the top of each graph
In parallel, we generated simulated graphs representing outcomes of the above experiment at different S → A and A → S conversion rates as in (Jeffery et al. 2013). In these simulations, we assumed that each culture originates with one single cell and a one single conversion event could take place in any of the first six generations. We also assumed that the proportions of mini-cultures in which the seeding cell will have an active or silent HTB1-yEGFP gene, respectively, would be similar to the proportion of GFP + cells in the bulk seeding cultures. In Fig. 5A, we present a simulation of a strain with 30% GFP + cells that has grown for 8–11 generations at 9% A → S and 5% S → A conversion rates per generation. In Fig. 5B, we present a simulation of a strain that has grown for 8–11 generations at 5% A → S and 3% S → A conversion rates. In these experiments, the prevalence of colonies with similar close percentage of GFP-positive cells (as represented by similar height of the multiple bars) reflect higher rate of epigenetic conversions. The appearance of lower or higher bars at the flanks of the graphs indicates a reduced rate of epigenetic conversions.
In Fig. 5C–H, we present the graphs produced by our single and double deletion mutants. Based on the simulation in Fig. 5A, in the BY4742 strain, the conversion rates of the HTB1-yEGFP construct are approximately 9% A → S and 5% S → A per generation (Fig. 5C). The plot produced by the cac1∆ strain is consistent with a decrease in the S → A conversions at this locus (Fig. 5D). For example, out of 36 mini-cultures, three showed less than 10% yEGFP+ cells, while the proportion of GFP+ cells in the seeding culture was 63%. A reduction in the A → S conversions can also explain the abundance of mini-cultures with high proportion of GFP+ cells (Fig. 5D). The plot produced by the asf1∆ strain was similar to the plot produced by BY4742 with some indication of reduced A → S conversions (Fig. 5E). The plot produced by the tof1∆ strain indicated a reduction in the overall frequency of conversion rates, but less pronounced as compared to the cac1∆ strain (Fig. 5F). In the tof1∆cac1∆ strain, the distribution of yEGFP+ cells in the plot indicated conversion rates higher than in the BY4742 strain but lower than the cac1∆ strain (Fig. 5G). Hence, these observations support the idea that the reduction of conversion rates in cac1∆ is reversed by the deletion of TOF1. Similar effect was observed at the HMLα, but not the FLO11 locus. Interestingly, the deletion of TOF1 in the asf1∆ background produced a plot similar to the one observed in the cac1∆ strain while neither tof1∆ nor asf1∆ strains displayed a similarly strong phenotype (Fig. 5H). This outcome is consistent with the notion that the deletion of TOF1 can exacerbate the silencing deficiency of asf1∆ and that these effects can also be linked to the conversion rates at this locus.
Discussion
The mechanisms of silencing at the mating type, the sub-telomeric, the FLO genes, and the rRNA-array loci are well understood (Gartenberg and Smith 2016; Shaban et al. 2021), but little is known on how the passage of replication forks affect the epigenetic state and epigenetic conversions in them (Rowlands et al. 2017; Stewart-Morgan et al. 2020). In this study, we addressed this question by combinations of deletions in genes that encode histone chaperones engaged in replication-coupled chromatin reassembly (CAC1 and ASF1) and factors that work at paused replication forks (TOF1 and RRM3) (Stewart-Morgan et al. 2020). CAC1 encodes a subunit of CAF-1, which travels behind the fork through an association with the replication clamp PCNA (POL30), while Asf1p is believed to travel ahead of the fork. TOF1 encodes a component of the FPC, while RRM3 encodes a DNA helicase believed to rescue stalled forks (Shyian and Shore 2021). Recent structural studies indicate that Tof1p/Csm3p associate with the fork ahead of the MCM helicase (Baretić et al. 2020). Because Rrm3p interacts with PCNA, it is believed that it travels behind the fork (Schmidt et al. 2002).
TOF1 and RRM3 regulate gene silencing in conjunction with CAF-1 and ASF1
In this study, we found that the deletion of TOF1 reduced the loss of silencing in cac1∆ cells but enhanced the loss of silencing in asf1∆ cells at both the VIIL telomere and HMLα (Figs. 2 and 4). The deletion of RRM3 in both cac1∆ and asf1∆ cells reduced silencing. Hence, we demonstrate that TOF1 (and potentially the FPC) and RRM3 are involved in gene silencing and that their functions are connected to the functions of the replication-coupled chaperones CAF-1 and ASF1. We have not observed similar loss of silencing in cells harboring a deletion of PIF1. PIF1 encodes a DNA helicase homologous to RRM3 and is known to promote DNA replication through G-quadruplex motifs on DNA (Pohl and Zakian 2019; Paeschke et al. 2011). On the other hand, RRM3 is necessary for rescuing paused replication forks at positions of tightly bound DNA proteins (Sauty et al. 2021). While it is preliminary to address if G-quadruplexes affect gene silencing, our current results and the known activities of TOF1 and RRM3 suggest that the mechanisms behind the observed genetic interactions operate at replication forks paused at positions of tightly bound proteins.
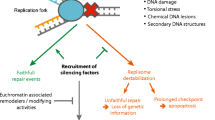
It is established that Tof1p directly interacts with Topoisomerase I (Park and Sternglanz 1999) and that this interaction could be significant in the regulation of fork pausing (Shyian et al. 2020). It has also been shown that amino acids 762–830 of Tof1p are critical for the pausing of the replication forks and for sensitivity to DNA damage (Westhorpe et al. 2020). In Fig. 2E, we have demonstrated that the deletion of this portion of Tof1p leads to substantial loss of silencing at the telomeres but does not completely phenocopy the loss of TOF1. These results strongly suggest that the pausing of the fork has a major effect on the silencing of genes at the sub-telomeres. At the same time, the differences in the effects of the deletion TOF1 and the tof1-762∆ truncation indicate a more complex mechanism. For example, it is not clear to what extent these truncations affect the interaction with Top1p. The exploration of the physical interaction between Tof1p and FACT (Safaric et al. 2022) via genetic analyses is also of interest. As for RRM3, apart from its physical interaction with PCNA (Schmidt et al. 2002), little is known about its mode of action. In Fig. 6, we present a model that summarizes our findings and the possible mechanism that can lead to the observed effects.

A model for the interaction between histone chaperones, FPC and Rrm3p at paused replication forks
Previous studies have indicated that CAF-1 and ASF1 function in distinct genetic pathways at the mating type loci and the telomeres. For example, loss of gene silencing at the telomeres and HML locus is exacerbated by the deletion of the histone chaperone Rtt106 in cac1∆ but not asf1∆ cells (Sharp et al. 2001; Jeffery et al. 2013; Janke et al. 2018). On the other hand, the deletion of clamp loader ELG1 reduces the silencing at HML in asf1∆ but not cac1∆ cells (Janke et al. 2018). Our findings support the notion of the functional and mechanistic distinction between these two histone chaperones. Specifically, we demonstrate that the deletion of TOF1 has opposite effects in cac1∆ and asf1∆ cells. It is possible that these differential effects are linked to the physical proximity of FPC and ASF1 ahead of the fork and the distortion of the helicase-polymerase conformation upon pausing of the replisome. In addition, the deletions of RRM3 and TOF1 have opposite effects on the stability of the paused forks at the rRNA gene array (Mohanty et al. 2006; Bastia et al. 2016) but seem to act independently of each other (Shyian et al. 2020). In earlier studies (Wyse et al. 2016; Rowlands et al. 2019b) and here we have shown that the deletion of RRM3 in both cac1∆ and asf1∆ backgrounds leads to significant loss of silencing at all loci tested. On the other hand, our assays in the rrm3∆tof1∆ strain did not conclusively show opposite gene silencing activities of these two factors. Further studies are needed to elucidate the mechanisms behind the genetic interactions of these genes.
TOF1 is not involved in the silencing of the FLO11 locus
Here, we show that the effects of the deletion of TOF1 at the VIIL telomere and HMLα are not recaptured at FLO11 (Fig. 3). The silencing of FLO11 is achieved through an SIR-independent mechanism while the silencing at telomeres and HMLα is SIR-dependent (Rowlands et al. 2017; Sauty et al. 2021). It is possible that Tof1p communicates with SIR proteins to maintain the silent state. Nevertheless, we favor the idea that forks do not pause at the FLO loci. Indeed, fork pausing was not detected at these positions in genome-wide studies in wild-type and rrm3∆ strains (Ivessa et al. 2003; Azvolinsky et al. 2006). However, if this is true, we need to consider that the effect of the deletion of RRM3 at the FLO11 locus (Rowlands et al. 2019b) (Fig. 3) is not caused by its role in the pausing of the fork, but by another, yet uncovered function of this helicase. Support for this idea comes from the fact that Rrm3p is associated with elongating forks and is required for overall fork processivity in vivo (Azvolinsky et al. 2006). It is possible that Mrc1p is also involved in sensing pausing and in the maintenance of gene silencing. However, we could not address this question in our system, because the deletion of MRC1 was lethal in conjunction with the deletions of CAC1 and ASF1.
The frequencies of epigenetic conversions reflect the silencing phenotypes of cac1∆tof1∆ and asf1∆tof1∆
We conducted extensive analyses of the frequencies of S → A and A → S conversions at the VIIL locus. In tune with our earlier study (Jeffery et al. 2013), we demonstrate that the loss of silencing in cac1∆ cells is linked to the reduction of the frequency of both S → A and A → S conversions (Fig. 5D). These observations agree with the idea that CAF-1 is engaged in the assembly of H3/H4 tetramers from “new” histones only (Ahmad and Henikoff 2018). If this is the case, at paused replication forks the supply of “old” histones could be reduced thus allowing the deposition of “new” histones and loss of epigenetic marks. The removal of CAF-1 would reduce the deposition of new histones and a greater reduction in S → A rates than the A → S rates will lead to the accumulation of GFP + cells in the culture.
Why does the deletion of TOF1 reverse this effect? A plausible explanation is Tof1p (and FPC), while stabilizing the paused replisome, also prevents spurious deposition or exchange of histones on the new DNA strands. This activity of FPC could include ASF1, but also other histone chaperones. For example, recent studies have indicated that Tof1p physically interacts with FACT (Safaric et al. 2022) and that efficient replisome progression on chromatin templates in vitro requires FACT (Kurat et al. 2017). On the other hand, it has been suggested that Asf1p, apart from its role in the disassembly of nucleosomes, can also retain the “old” histones in the vicinity of paused forks and supply them upon resumption of elongation (reviewed in (Alabert and Groth 2012). FACT could have a similar role in the retention of H2A/H2B histones at the paused replisome. When this function is lost in asf1∆ cells, FPC prevents the deposition of new histones thus contributing to the stability of epigenetic transmission. When both asf1 and tof1 are lost, the loss of “old” histones is exacerbated, and epigenetic stability diminishes.
DDK, CAF-1, and Tof1p
Another possibility for the epistatic interaction of CAC1 and TOF1 is the involvement of a third factor. It is tempting to speculate that this factor is the Dbf4-Dependent Kinase, DDK. The Cac1p subunit of CAF-1 can be phosphorylated by DDK in both human and budding yeast cell extracts (Gerard et al. 2006; Jeffery et al. 2015) and mutations of the DDK target sites on the yeast CAC1 lead to loss of silencing (Jeffery et al. 2015; Rowlands et al. 2019a). Tof1p is also phosphorylated upon pausing of the fork and its phosphorylation is linked to the stability of paused replisome (Bastia et al. 2016). However, it is not clear if DDK is the kinase responsible for this effect (Bastia et al. 2016). Still, it is possible that the phosphorylation of these two substrates is coordinated at the paused replisome and the loss of one can affect the activity of the other.
Variations in the outcomes of different gene silencing assays
The recently introduced CRASH assay is remarkably sensitive and detects minor transient loss of silencing at HMLα that previous URA3/5-FOA assays cannot capture (Janke et al. 2018; Brothers and Rine 2019). On the other hand, our HTB1-yEGFP reporter assay is less sensitive than the URA3/5-FOA assay (Shaban et al., submitted). It seems that the exposure to 5-FOA detects levels of expression of URA3, which, like in the CRASH assays, represent transient loss of silencing at the telomere and not necessarily true epigenetic conversions. Still, our drug-free assay reveals the same trends observed with the URA3/5-FOA assays, albeit in significantly narrower range. More importantly, the fact that we see the same genetic interactions with the most (CRASH) and the least (HTB1-yEGFP) sensitive assays adds credibility to our analyses and conclusions.
Conclusion
Our findings provide strong evidence about the involvement of TOF1 (and possibly the FPC) and RRM3 in maintenance of epigenetic state through interactions with the replication-coupled histone chaperones CAF-1 and ASF1. Future studies are needed to add detailed evidence for the mechanisms that govern histone turnover at paused replication forks.
Change history
24 November 2023
A Correction to this paper has been published: https://doi.org/10.1007/s00294-023-01279-x
References
Ahmad K, Henikoff S (2018) No strand left behind. Science 361:1311–1312. https://doi.org/10.1126/science.aav0871
Alabert C, Groth A (2012) Chromatin replication and epigenome maintenance. Nat Rev Mol Cell Biol 13:153–167. https://doi.org/10.1038/nrm3288
Almouzni G, Cedar H (2016) Maintenance of epigenetic information. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a019372
Azvolinsky A, Dunaway S, Torres JZ et al (2006) The S. cerevisiae Rrm3p DNA helicase moves with the replication fork and affects replication of all yeast chromosomes. Genes Dev 20:3104–3116. https://doi.org/10.1101/gad.1478906
Baretić D, Jenkyn-Bedford M, Aria V et al (2020) Cryo-EM structure of the fork protection complex bound to CMG at a replication fork. Mol Cell 78:926-940.e13. https://doi.org/10.1016/j.molcel.2020.04.012
Bastia D, Srivastava P, Zaman S et al (2016) Phosphorylation of CMG helicase and Tof1 is required for programmed fork arrest. Proc Natl Acad Sci. https://doi.org/10.1073/pnas.1607552113
Beranek DT, Heflich RH, Kodell RL, Morris SM, Casciano DA (1983) Correlation between specific DNA-methylation products and mutation induction at the HGPRT locus in Chinese hamster ovary cells. Mutat Res 110:171–180. https://doi.org/10.1016/0027-5107(83)90026-x
Brothers M, Rine J (2019) Mutations in the PCNA DNA polymerase clamp of saccharomyces cerevisiae reveal complexities of the cell cycle and ploidy on heterochromatin assembly. Genetics 213:449–463. https://doi.org/10.1534/genetics.119.302452
Deegan TD, Baxter J, Ortiz Bazán MÁ et al (2019) Pif1-family helicases support fork convergence during DNA replication termination in eukaryotes. Mol Cell 74:231-244.e9. https://doi.org/10.1016/j.molcel.2019.01.040
Dodson AE, Rine J (2015) Heritable capture of heterochromatin dynamics in Saccharomyces cerevisiae. Elife 4:e05007–e05007. https://doi.org/10.7554/eLife.05007
Gan H, Serra-Cardona A, Hua X et al (2018) The Mcm2-Ctf4-Polα axis facilitates parental histone H3–H4 transfer to lagging strands. Mol Cell 72:140-151.e3. https://doi.org/10.1016/j.molcel.2018.09.001
Gartenberg MR, Smith JS (2016) The Nuts and Bolts of Transcriptionally Silent Chromatin in Saccharomyces cerevisiae. Genetics 203:1563–1599. https://doi.org/10.1534/genetics.112.145243
Gerard A, Koundrioukoff S, Ramillon V et al (2006) The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO Rep 7:817–823. https://doi.org/10.1038/sj.embor.7400750
Gottschling DE, Aparicio OM, Billington BL, Zakian VA (1990) Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63:751–762
Groth A, Corpet A, Cook AJ et al (2007) Regulation of replication fork progression through histone supply and demand. Science 318:1928–1931. https://doi.org/10.1126/science.1148992
Hastie T, Tibshirani R, Friedman J (2009) The elements of statistical learning. Springer Ser Statist. https://doi.org/10.1007/978-0-387-84858-7
Ivessa AS, Lenzmeier BA, Bessler JB et al (2003) The Saccharomyces cerevisiae helicase Rrm3p facilitates replication past nonhistone protein-DNA complexes. Mol Cell 12:1525–1536
Janke R, King GA, Kupiec M, Rine J (2018) Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc Natl Acad Sci U S A 115:E2030–E2039. https://doi.org/10.1073/pnas.1721573115
Jeffery DC, Wyse BA, Rehman MA et al (2013) Analysis of epigenetic stability and conversions in Saccharomyces cerevisiae reveals a novel role of CAF-I in position-effect variegation. Nucleic Acids Res 41:8475–8488. https://doi.org/10.1093/nar/gkt623
Jeffery DC, Kakusho N, You Z et al (2015) CDC28 phosphorylates Cac1p and regulates the association of chromatin assembly factor I with chromatin. Cell Cycle 14:74–85. https://doi.org/10.4161/15384101.2014.973745
Kurat CF, Yeeles JTP, Patel H, Early A, Diffley JFX (2017) Chromatin controls DNA replication origin selection, lagging-strand synthesis, and replication fork rates. Mol Cell 65:117–130. https://doi.org/10.1016/j.molcel.2016.11.016
Makovets S, Herskowitz I, Blackburn EH (2004) Anatomy and dynamics of DNA replication fork movement in yeast telomeric regions. Mol Cell Biol 24:4019–4031
Mano Y, Kobayashi TJ, Nakayama J et al (2013) Single cell visualization of yeast gene expression shows correlation of epigenetic switching between multiple heterochromatic regions through multiple generations. PLoS Biol 11:e1001601–e1001601. https://doi.org/10.1371/journal.pbio.1001601
Mohanty BK, Bairwa NK, Bastia D (2006) The Tof1p-Csm3p protein complex counteracts the Rrm3p helicase to control replication termination of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 103:897–902. https://doi.org/10.1073/pnas.0506540103
Paeschke K, Capra JA, Zakian VA (2011) DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145:678–691. https://doi.org/10.1016/j.cell.2011.04.015
Park H, Sternglanz R (1999) Identification and characterization of the genes for two topoisomerase I-interacting proteins from Saccharomyces cerevisiae. Yeast 15:35–41. https://doi.org/10.1002/(SICI)1097-0061(19990115)15:1%3c35::AID-YEA340%3e3.0.CO;2-R
Petryk N, Dalby M, Wenger A et al (2018) MCM2 promotes symmetric inheritance of modified histones during DNA replication. Science 361:1389–1392. https://doi.org/10.1126/science.aau0294
Pohl TJ, Zakian VA (2019) Pif1 family DNA helicases: a helpmate to RNase H? DNA Repair 84:102633. https://doi.org/10.1016/j.dnarep.2019.06.004
Rossmann MP, Luo W, Tsaponina O et al (2011) A common telomeric gene silencing assay is affected by nucleotide metabolism. Mol Cell. https://doi.org/10.1016/j.molcel.2011.03.007
Rowlands H, Dhavarasa P, Cheng A, Yankulov K (2017) Forks on the run: can the stalling of DNA replication promote epigenetic changes? Front Genet 8:86. https://doi.org/10.3389/fgene.2017.00086
Rowlands H, Shaban K, Cheng A et al (2019a) Dysfunctional CAF-I reveals its role in cell cycle progression and differential regulation of gene silencing. Cell Cycle 18:3223–3236. https://doi.org/10.1080/15384101.2019.1673100
Rowlands H, Shaban K, Foster B et al (2019b) Histone chaperones and the Rrm3p helicase regulate flocculation in S. cerevisiae. Epigenet Chromatin 12:56. https://doi.org/10.1186/s13072-019-0303-8
Rusche LN, Kirchmaier AL, Rine J (2003) The establishment, inheritance, and function of silenced chromatin in Saccharomyces cerevisiae. Annu Rev Biochem 72:481–516. https://doi.org/10.1146/annurev.biochem.72.121801.161547
Safaric B, Chacin E, Scherr MJ et al (2022) The fork protection complex recruits FACT to reorganize nucleosomes during replication. Nucleic Acids Res 50:1317–1334. https://doi.org/10.1093/nar/gkac005
Sauty SM, Shaban K, Yankulov K (2021) Gene repression in S. cerevisiae—looking beyond Sir-dependent gene silencing. Curr Genet 67:3–17. https://doi.org/10.1007/s00294-020-01114-7
Schmidt KH, Derry KL, Kolodner RD (2002) Saccharomyces cerevisiae RRM3, a 5′ to 3′ DNA helicase, physically interacts with proliferating cell nuclear antigen. J Biol Chem 277:45331–45337. https://doi.org/10.1074/jbc.M207263200
Scully R, Elango R, Panday A et al (2021) Recombination and restart at blocked replication forks. Curr Opin Genet Dev 71:154–162. https://doi.org/10.1016/j.gde.2021.08.003
Shaban K, Sauty SM, Yankulov K (2021) Variation, variegation and heritable gene repression in S. cerevisiae. Front Genet 12:630506. https://doi.org/10.3389/fgene.2021.630506
Shaban K, Sauty SM, Fisher A et al (2023) Evaluation of drug-free methods for the detection of gene silencing in Saccharomyces cerevisiae. Biochem Cell Biol 101:125–130. https://doi.org/10.1139/bcb-2022-0243
Sharp JA, Fouts ET, Krawitz DC, Kaufman PD (2001) Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr Biol 11:463–473. https://doi.org/10.1016/S0960-9822(01)00140-3
Shyian M, Shore D (2021) Approaching protein barriers: emerging mechanisms of replication pausing in eukaryotes. Front Cell Dev Biol 9:672510. https://doi.org/10.3389/fcell.2021.672510
Shyian M, Albert B, Zupan AM et al (2020) Fork pausing complex engages topoisomerases at the replisome. Genes Dev 34:87–98. https://doi.org/10.1101/gad.331868.119
Stewart-Morgan KR, Petryk N, Groth A (2020) Chromatin replication and epigenetic cell memory. Nat Cell Biol 22:361–371. https://doi.org/10.1038/s41556-020-0487-y
Sutton A, Bucaria J, Osley MA, Sternglanz R (2001) Yeast ASF1 protein is required for cell cycle regulation of histone gene transcription. Genetics 158:587–596. https://doi.org/10.1093/genetics/158.2.587
Takahashi YH, Schulze JM, Jackson J et al (2011) Dot1 and histone H3K79 methylation in natural telomeric and HM silencing. Mol Cell 42:118–126. https://doi.org/10.1016/j.molcel.2011.03.006
Tyler JK, Adams CR, Chen SR et al (1999) The RCAF complex mediates chromatin assembly during DNA replication and repair. Nature 402:555–560. https://doi.org/10.1038/990147
Westhorpe R, Keszthelyi A, Minchell NE et al (2020) Separable functions of Tof1/Timeless in intra-S-checkpoint signalling, replisome stability and DNA topological stress. Nucleic Acids Res 48:12169–12187. https://doi.org/10.1093/nar/gkaa963
Wyse B, Oshidari R, Rowlands H et al (2016) RRM3 regulates epigenetic conversions in Saccharomyces cerevisiae in conjunction with chromatin assembly factor I. Nucleus 7:405–414. https://doi.org/10.1080/19491034.2016.1212796
Yankulov K (2013) Dynamics and stability: epigenetic conversions in position effect variegation. Biochem Cell Biol 91:6–13. https://doi.org/10.1139/bcb-2012-0048
Yeeles JTP, Janska A, Early A, Diffley JFX (2017) How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65:105–116. https://doi.org/10.1016/j.molcel.2016.11.017
Yu C, Gan H, Serra-Cardona A et al (2018) A mechanism for preventing asymmetric histone segregation onto replicating DNA strands. Science 361:1386–1389. https://doi.org/10.1126/science.aat8849
Zhang Z, Shibahara K, Stillman B (2000) PCNA connects DNA replication to epigenetic inheritance in yeast. Nature 408:221–225. https://doi.org/10.1038/35041601
Zunder RM, Rine J (2012) Direct interplay among histones, histone chaperones, and a chromatin boundary protein in the control of histone gene expression. Mol Cell Biol 32:4337–4349. https://doi.org/10.1128/MCB.00871-12
Acknowledgements
The authors would like to thank Dr. M. Oki for providing the HTB1-yEGFP construct, Dr. H. Murphy for FLO11-yGFP-KanMX construct, and Dr. Jasper Rine for the CRASH strains, Emma Lessard for help in generating the mutant strains with the HTB1-yEGFP construct, the Molecular and Cellular Imaging Facility, University of Guelph for the technical support.
Funding
Funding for this study is provided by a grant to KY (RGPIN-2015-06727) from NSERC. KS, AD, AF and SMS are supported by bursaries from the College of Biological Science at the University of Guelph.
Author information
Authors and Affiliations
Contributions
KS planned and performed experiments, analyzed data, prepared figures and wrote a draft of the manuscript. AD performed experiments, analyzed data and prepared figures. SMS performed experiments and prepared figures. EL performed experiments and analyzed data. AF performed experiments and prepared figures. KY supervised the study and wrote the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by M. Polymenis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised due to correction in the Materials and methods section on page 3.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Shaban, K., Dolson, A., Fisher, A. et al. TOF1 and RRM3 reveal a link between gene silencing and the pausing of replication forks. Curr Genet 69, 235–249 (2023). https://doi.org/10.1007/s00294-023-01273-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-023-01273-3