
Abstract
Quiescence is a highly conserved inactive life stage in which the cell reversibly exits the cell cycle in response to external cues. Quiescence is essential for diverse processes such as the maintenance of adult stem cell stores, stress resistance, and longevity, and its misregulation has been implicated in cancer. Although the non-cycling nature of quiescent cells has made obtaining sufficient quantities of quiescent cells for study difficult, the development of a Saccharomyces cerevisiae model of quiescence has recently enabled detailed investigation into mechanisms underlying the quiescent state. Like their metazoan counterparts, quiescent budding yeast exhibit widespread transcriptional silencing and dramatic chromatin condensation. We have recently found that the structural maintenance of chromosomes (SMC) complex condensin binds throughout the quiescent budding yeast genome and induces the formation of large chromatin loop domains. In the absence of condensin, quiescent cell chromatin is decondensed and transcription is de-repressed. Here, we briefly discuss our findings in the larger context of the genome organization field.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
A genome-wide shift in chromatin structure reprograms transcription during quiescence
A subset of budding yeast cells enter quiescence once dextrose, ethanol, and other nutrient supplies have been exhausted from the media (Allen et al. 2006; De Virgilio, 2012; Galdieri et al. 2010; Gray et al. 2004). Quiescent yeasts undergo a number of physiological changes that confer extended stress resistance and longevity (Allen et al. 2006; Werner-Washburne et al. 1993). During quiescence entry, a small number of quiescence and stress response genes are activated and the majority of the genome is repressed (Galdieri et al. 2010; Kuang et al. 2017; Li et al. 2015; McKnight et al. 2015; Roche et al. 2017). As one of the conserved hallmarks of quiescence is chromatin condensation, an attractive hypothesis has been that compaction of the chromatin fiber leads to the widespread transcriptional silencing observed in quiescent cells (Evertts et al. 2013; Laporte et al. 2016; Lohr and Ide 1979; Ngubo et al. 2011; Pinon, 1978; Rawlings et al. 2011; Rutledge et al. 2015). Consistently, the volume of yeast chromatin measured by fluorescence microscopy decreases by approximately 40% during quiescence (Swygert et al. 2019). Nucleosome density also increases in quiescent yeast cells and histone acetylation modifications associated with open, active chromatin structure are lost almost entirely due to global targeting of the Rpd3 histone deacetylase complex (McKnight et al. 2015). However, determining the higher-order structure of chromatin within cells at sufficient resolution to establish the relationship between chromatin fiber structure and the transcriptional state of individual genes has been difficult in any organism (Hsieh et al. 2015). To address this problem, we recently took advantage of Micro-C XL, a variant of the chromatin conformation capture family of techniques that uses micrococcal nuclease in place of restriction endonucleases, to map chromatin structure in quiescent at single-nucleosome resolution (Hsieh et al. 2015, 2016; Swygert et al. 2019).
The first Micro-C study identified chromosomal interaction domains (CIDs) in exponentially growing (log) budding yeast, similar to topologically associated domains (TADs) in metazoans. CIDs are 0.5–8 kilobase (about 2 kilobase on average) regions of the genome in which chromatin interactions occur frequently, separated by boundaries across which chromatin interactions are rare (Hsieh et al. 2015). Like their counterparts in higher organisms, yeast chromatin domains exist in nested form, in which CIDs may preferentially interact with other neighboring CIDs forming looser larger domains (Dixon et al. 2012; Hsieh et al. 2015, 2016). The positions of chromatin domain boundaries are largely conserved between log and quiescent cells (Swygert et al. 2019). However, in quiescent cells, chromatin interactions across CID clusters representing domains on the order of 10–60 kilobases (about 13 kilobases on average) increase (Swygert et al. 2019). To distinguish these larger domains from CIDs, we refer to them as large-CIDs (L-CIDs). While L-CIDs appear assembled in log cells, chromatin interactions within L-CIDs occur at longer distances in quiescent cells and the boundaries between L-CIDs are more insulating (Swygert et al. 2019). Strikingly, only in quiescent cells, we observed off-diagonal “corner” interactions between L-CID boundaries that indicate L-CIDs in quiescence are stabilized by the formation of discrete chromatin loops (Rao et al. 2014, 2017; Swygert et al. 2019).
A decade of intense investigation has revealed that chromatin domains are frequently stabilized by SMC complexes, large ring-shaped protein complexes that are believed to organize the genome by the movement of chromatin through their central cavity (Dixon et al. 2012; Hassler et al. 2018; Kakui and Uhlmann 2018; Rao et al. 2014. 2017; Yuen and Gerton 2018). Intriguingly, we found that cohesin, which is most commonly associated with TAD boundaries, transitions from binding throughout the log cell genome to being almost entirely absent from quiescent cell chromatin (Swygert et al. 2019). In contrast, condensin, which during log is largely restricted to centromeres, tRNA genes, and the rDNA, binds the vast majority of L-CID boundaries in quiescent cells (Swygert et al. 2019). The majority of condensin sites occur at nucleosome depleted regions (NDRs) at the promoters of RNA polymerase-II (Pol-II) occupied genes, primarily falling at intergenic regions between divergently oriented genes, although many peaks also fall between tandemly oriented genes. Condensin depletion during quiescence entry leads to the loss of approximately half of L-CID boundaries, a decrease in chromatin interactions within L-CIDs, and a substantial reduction in long-range looping (Swygert et al. 2019). Furthermore, even a partial loss of condensin binding during quiescence leads to an increase in chromatin volume to log levels (Swygert et al. 2019).
To determine the effect of condensin depletion on transcription, we performed ChIP-seq of Pol-II in wild-type and condensin-depleted quiescent cells. In this context, ChIP-seq measurement of Pol-II occupancy over gene bodies has two major advantages over RNA-seq measurements of steady-state mRNA levels. First, during quiescence entry, many mRNAs are stabilized in processing bodies as a response to stress (Aragon et al. 2006; Young et al. 2017). As a result, RNA-seq can misrepresent the identity and activity of genes that are transcribed during quiescence. Second, a recent study in which condensin was depleted from the chromatin of log cells found that condensin loss during replication can lead to the mis-segregation of the exosome, resulting in an increase in steady-state RNA levels without changes in transcription (Hocquet et al. 2018; Paul et al. 2018, 2019). Our data show that upon condensin depletion, Pol-II peaks increase in number corresponding to de-repression of approximately 20% of the entire genome (Swygert et al. 2019). The genes with the largest increases in Pol-II occupancy occur directly adjacent to the strongest condensin ChIP-seq peaks (Swygert et al. 2019). However, many genes located tens of kilobases from condensin binding sites are also activated, suggesting condensin may work both through action at gene promoters and at a distance.
How does condensin repress transcription during quiescence?
A common theme in the study of quiescence is that proteins and complexes such as Rpd3 that play relatively small roles in log perform crucial specialized functions in quiescent cells (McKnight et al. 2015). Similarly, the ability of condensin to regulate transcription in budding yeast is specific to its function at the L-CID boundaries during quiescence, as two recent studies have demonstrated that condensin depletion during log does not affect transcription (Hocquet et al. 2018; Paul et al. 2018). However, the mechanism of transcriptional repression in this context is not clear. In metazoans, the current model of TAD function in transcription is that TADs facilitate interactions between enhancers in promoters within the same TAD and insulate against interactions between enhancers and promoters in different TADs (Deng et al. 2012; Hansen et al. 2018; Lupianez et al. 2015; Nora et al. 2012). Consequently, although the disruption of TAD boundaries can alter the transcription of individual genes leading to deleterious effects, cohesin depletion leading to loss of all chromatin loop domains in mammalian cells had relatively modest effects on transcription genome-wide (Rao et al. 2017). In contrast, enhancer elements capable of regulating gene expression at long distances have not been identified in budding yeast, and most genes are regulated by sequences within a kilobase of their transcription start sites (Bulger and Groudine 2011).
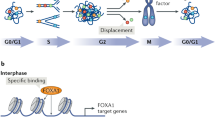
We have proposed a model in which condensin represses transcription in quiescent yeast cells by two mechanisms (Fig. 1). First, condensin binding at the promoters of active genes prevents aberrant activation of divergently oriented genes whose promoters share the same intergenic region. Second, the increase in chromatin interactions within L-CIDs in quiescence sterically blocks binding of transcriptional activators to genes located with L-CID loops. This model accounts for how condensin prevents activation of genes both nearby and at a distance. Importantly, though condensin loss leads to chromatin decompaction genome-wide, this change in chromatin structure is not sufficient to activate transcription as not all genes are de-repressed upon condensin depletion, including a subset of genes located near condensin binding sites (Swygert et al. 2019). This suggests that higher-order chromatin structure functions to control the accessibility of genes to transcription factors and polymerases, but that chromatin compaction state alone does not dictate transcriptional activity.

Model of condensin-dependent transcriptional silencing. During quiescence, condensin loaded onto chromatin extrudes a loop until blocked by a transcription factor (TF) or by transcription itself. This process forms an L-CID with condensin bound at boundaries on either side. Condensin’s presence insulates genes across boundaries from inappropriate transcriptional activation and promotes chromatin compaction interactions within L-CIDs that sterically inhibit binding of transcription factors and Pol-II
How are chromatin domain boundaries defined in S. cerevisiae?
Condensin transitions from binding relatively few highly specialized genomic loci in log cells to binding L-CID boundaries at gene promoters throughout the genome in quiescent cells through an unknown mechanism. The most highly supported model of TAD formation is that an SMC complex is loaded onto a loop of chromatin either as a monomer so that one portion of chromatin passes through the SMC ring while the other portion is bound by the head domain comprising the kleisin and heat-repeat containing subunits (as in Fig. 1), or as a dimer so that each end of the chromatin loop passes through an SMC ring (Eeftens et al. 2017; Ganji et al. 2018; Hassler et al. 2018; Keenholtz et al. 2017; Matityahu and Onn, 2018; Wang et al. 2018). Chromatin is then actively or passively extruded through the SMC ring until a barrier is encountered that blocks further extrusion through the loop (Eeftens et al. 2017; Fudenberg et al. 2017; Hassler et al. 2018; Terakawa et al. 2017; Wang et al. 2018). In vertebrates, this barrier is commonly the multiple zinc finger transcription factor CTCF, which flanks a large proportion of TAD boundaries in a convergent orientation (de Wit et al. 2015; Dixon et al. 2012; Sanborn et al. 2015). However, no architectural protein similar to CTCF has been identified in yeast.
In Schizosaccharomyces pombe, condensin localizes to actively transcribed genes during mitosis bound by the transcription factors Ace2 and Ams2 (Kim et al. 2016; Robellet et al. 2017). These transcription factors are capable of physical interaction with condensin, and insertion of ectopic sites for these factors leads to condensin recruitment in vivo (Kim et al. 2016). Interestingly, condensin in mitotic S. pombe localizes both to the 5′ and 3′ ends of active genes and in mitotic human cells “bookmarks” highly active interphase genes, suggesting a combination of transcription factors and transcription itself may be involved in regulating condensin loop extrusion (Hocquet et al. 2018; Kim et al. 2016; Sutani et al. 2015; Tanizawa et al. 2017; Toselli-Mollereau et al. 2016). Similarly, TFIIIC physically interacts with condensin and plays a role in condensin recruitment to RNA polymerase-III-transcribed tRNA genes in yeasts and to a broader class of TFIIIC-bound genes in mammals (D’Ambrosio et al. 2008; Haeusler et al. 2008; Yuen and Gerton, 2018; Yuen et al. 2017). What role TFIIIC may play in quiescence, during which condensin appears to leave tRNA genes in favor of Pol-II transcribed promoters, is currently unclear.
During quiescence entry, the transcriptional activator Msn2 and the partially redundant Msn4 enter the nucleus and bind the stress response element sequence at stress response gene promoters (Estruch 2000; Kuang et al. 2017, 2018; Li et al. 2015). Consequently, the majority of actively transcribed genes in quiescent cells are regulated by Msn2, and there is a high degree of overlap between Msn2 and condensin binding sites (Swygert et al. 2019). Although this suggests Msn2 may be involved in recruiting condensin to L-CID boundaries, deletion of the Msn2 gene reduces but does not abrogate condensin binding. Interestingly, although Msn2 is believed to be a key activator of quiescence-specific genes, deletion of Msn2 modestly increases Pol-II binding at both Msn2 targets and some condensin-regulated genes (Swygert et al. 2019). It is possible that this effect is due to a combination of partial redundancy by Msn4, which may fill some transcription gaps in place of Msn2, and a partial loss of condensin binding, which likely leads to an overall weakening of L-CID boundaries and a loss of L-CID boundaries in a subset of cells. However, one intriguing possibility is that transcription itself may play a role in targeting condensin. Active transcription has been shown to block cohesin loop extrusion and even to relocate cohesin molecules to the 3′ ends of genes by a seemingly passive mechanism (Busslinger et al. 2017; Glynn et al. 2004; Gullerova and Proudfoot 2008; Lengronne et al. 2004; Ocampo-Hafalla et al. 2016). Similarly, transcription slows but does not entirely prevent active loop extrusion by a bacterial condensin complex (Wang et al. 2018), perhaps by inducing an array of difficult-to-transverse supercoils. The seemingly contradictory mechanism by which active transcription appears to recruit condensin to repress transcription is a topic of current investigation by our group.
One of the most puzzling aspects of our findings is that although L-CID boundaries require condensin during quiescence, the majority of L-CID boundaries are conserved between quiescent and log cells despite the total absence of condensin at these regions (Swygert et al. 2019). What could be determining L-CID boundaries in log? Cohesin loading factor Scc2 localizes to many CID boundaries during log and cohesin has been found to partially regulate chromatin globule formation in S. pombe (Hsieh et al. 2015; Mizuguchi et al. 2014). However, transcription appears to “push” cohesin along chromatin so that in yeast it accumulates at the 3′ ends of genes and especially at intergenic regions between convergently transcribed genes (Glynn et al. 2004; Gullerova and Proudfoot 2008; Lengronne et al. 2004; Litwin and Wysocki 2018; Ocampo-Hafalla et al. 2016). Consequently, although cohesin may play a role in defining CID boundaries, L-CID boundaries, which overlap with the strongest CID boundaries at primarily divergently oriented genes, seem to exclude cohesin (Fig. 2a). Similarly, Smc5/6 is absent in the majority of L-CID boundaries in log (Swygert et al. 2019). A recent study has found that nucleosomes excluding DNA sequences are capable of insulating interactions between chromatin domains (Matsushima et al. 2019). Interestingly, the sites of condensin binding in quiescence fall at remarkably broad and deep NDRs in both quiescence and log (Fig. 2b, c). The persistence of large NDRs at L-CID boundaries in log cells exists despite the transcriptional inactivity of most quiescence-specific genes (Swygert et al. 2019). Whether the DNA sequence itself is generating these large NDRs or if an unknown factor maintains NDRs at L-CID boundaries in log is entirely unknown. Perhaps, a chromatin remodeling enzyme maintains an open NDR at stress response elements to facilitate rapid response to stresses such as heat shock. However, whether NDR maintenance is sufficient to create a boundary between otherwise loosely crumpled chromatin regions in log cells remains to be determined.

All data are from Swygert et al. 2019
L-CID boundaries persist in log cells despite the absence of SMC complex binding. a Condensin and cohesin ChIP-seq data at transcription start sites at quiescent L-CID boundaries (TSSs) in log and quiescent cells. b, c Histone H3 ChIP-seq data at quiescent L-CID boundary TSSs and all remaining TSSs (other) in log (b) and quiescent (c) cells
Are L-CID boundaries clustered?
One of the known functions of condensin in yeast log cells is to cluster a subset of tRNA genes to the rDNA (Haeusler et al. 2008; Paul et al. 2018, 2019). This clustering occurs across chromosomes and thus must involve condensin dimerization or a cross-fiber method of condensin loading. Similarly, condensin is involved in clustering histone genes in mammals (Yuen et al. 2017). Consequently, it has been proposed that one of the functions of condensin may be to cluster highly expressed genes (Yuen and Gerton, 2018). In our Micro-C data, we have observed that interactions occur between both neighboring and distal L-CID boundaries in a nested fashion (Swygert et al. 2019). This could result from two non-exclusive mechanisms between which our data cannot distinguish. First, the loop extrusion model postulates that condensin complexes actively extrude chromatin until encountering a physical block. This means that depending on the site of condensin loading and the presence of transcription factors or other blocks in a given cell, the position of loops will likely sample all possible L-CID boundaries over a sufficiently large population of cells. However, a second possibility is that L-CID boundaries are able to interact in trans, perhaps through condensin dimerization or a related mechanism, leading to a three-dimensional clustering of L-CID boundaries. That the small number of actively transcribed genes in quiescent cells are clustered into transcription factories which both facilitate their transcription and prevent aberrant activation of nearby genes is an attractive possibility that warrants further investigation.
Future perspectives
Our discovery that the condensin complex relocates to the promoters of active genes in quiescent cells to form chromatin loop domains that repress transcription has provided novel insight into how widespread chromatin compaction functions to repress transcription during quiescence. To our knowledge, L-CIDs are the first SMC-dependent TAD-like chromatin domains discovered in budding yeast. The discovery of L-CIDs thus renders quiescent yeast a potentially vital model system for studying the mechanisms and functions of chromatin domains, but also raises important questions regarding the ubiquity of such domains in organisms lacking architectural factors such as CTCF and recognized distal modes of transcriptional regulation. Future investigation of the processes underlying quiescence is likely to continue to reveal surprising roles for both well-described and yet-unidentified factors.
References
Allen C, Buttner S, Aragon AD, Thomas JA, Meirelles O, Jaetao JE, Benn D, Ruby SW, Veenhuis M, Madeo F et al (2006) Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J Cell Biol 174:89–100
Aragon AD, Quinones GA, Thomas EV, Roy S, Werner-Washburne M (2006) Release of extraction-resistant mRNA in stationary phase Saccharomyces cerevisiae produces a massive increase in transcript abundance in response to stress. Genome Biol 7:R9
Bulger M, Groudine M (2011) Functional and mechanistic diversity of distal transcription enhancers. Cell 144:327–339
Busslinger GA, Stocsits RR, van der Lelij P, Axelsson E, Tedeschi A, Galjart N, Peters JM (2017) Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544:503–507
D’Ambrosio C, Schmidt CK, Katou Y, Kelly G, Itoh T, Shirahige K, Uhlmann F (2008) Identification of cis-acting sites for condensin loading onto budding yeast chromosomes. Genes Dev 22:2215–2227
De Virgilio C (2012) The essence of yeast quiescence. FEMS Microbiol Rev 36:306–339
de Wit E, Vos ES, Holwerda SJ, Valdes-Quezada C, Verstegen MJ, Teunissen H, Splinter E, Wijchers PJ, Krijger PH, de Laat W (2015) CTCF binding polarity determines chromatin looping. Mol Cell 60:676–684
Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA (2012) Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell 149:1233–1244
Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380
Eeftens JM, Bisht S, Kerssemakers J, Kschonsak M, Haering CH, Dekker C (2017) Real-time detection of condensin-driven DNA compaction reveals a multistep binding mechanism. EMBO J 36:3448–3457
Estruch F (2000) Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol Rev 24:469–486
Evertts AG, Manning AL, Wang X, Dyson NJ, Garcia BA, Coller HA (2013) H4K20 methylation regulates quiescence and chromatin compaction. Mol Biol Cell 24:3025–3037
Fudenberg G, Abdennur N, Imakaev M, Goloborodko A, Mirny LA (2017) Emerging evidence of chromosome folding by loop extrusion. Cold Spring Harb Symp Quant Biol 82:45–55
Galdieri L, Mehrotra S, Yu S, Vancura A (2010) Transcriptional regulation in yeast during diauxic shift and stationary phase. OMICS 14:629–638
Ganji M, Shaltiel IA, Bisht S, Kim E, Kalichava A, Haering CH, Dekker C (2018) Real-time imaging of DNA loop extrusion by condensin. Science 360:102–105
Glynn EF, Megee PC, Yu HG, Mistrot C, Unal E, Koshland DE, DeRisi JL, Gerton JL (2004) Genome-wide mapping of the cohesin complex in the yeast Saccharomyces cerevisiae. PLoS Biol 2:E259
Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M (2004) “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev 68:187–206
Gullerova M, Proudfoot NJ (2008) Cohesin complex promotes transcriptional termination between convergent genes in S. pombe. Cell 132:983–995
Haeusler RA, Pratt-Hyatt M, Good PD, Gipson TA, Engelke DR (2008) Clustering of yeast tRNA genes is mediated by specific association of condensin with tRNA gene transcription complexes. Genes Dev 22:2204–2214
Hansen AS, Cattoglio C, Darzacq X, Tjian R (2018) Recent evidence that TADs and chromatin loops are dynamic structures. Nucleus 9:20–32
Hassler M, Shaltiel IA, Haering CH (2018) Towards a unified model of SMC complex function. Curr Biol 28:R1266–R1281
Hocquet C, Robellet X, Modolo L, Sun XM, Burny C, Cuylen-Haering S, Toselli E, Clauder-Munster S, Steinmetz L, Haering CH et al (2018) Condensin controls cellular RNA levels through the accurate segregation of chromosomes instead of directly regulating transcription. Elife 7:e38517
Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, Rando OJ (2015) Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162:108–119
Hsieh TS, Fudenberg G, Goloborodko A, Rando OJ (2016) Micro-C XL: assaying chromosome conformation from the nucleosome to the entire genome. Nat Methods 13:1009–1011
Kakui Y, Uhlmann F (2018) SMC complexes orchestrate the mitotic chromatin interaction landscape. Curr Genet 64:335–339
Keenholtz RA, Dhanaraman T, Palou R, Yu J, D’Amours D, Marko JF (2017) Oligomerization and ATP stimulate condensin-mediated DNA compaction. Sci Rep 7:14279
Kim KD, Tanizawa H, Iwasaki O, Noma K (2016) Transcription factors mediate condensin recruitment and global chromosomal organization in fission yeast. Nat Genet 48:1242–1252
Kuang Z, Pinglay S, Ji H, Boeke JD (2017) Msn2/4 regulate expression of glycolytic enzymes and control transition from quiescence to growth. Elife 6:e29938
Kuang Z, Ji H, Boeke JD (2018) Stress response factors drive regrowth of quiescent cells. Curr Genet 64:807–810
Laporte D, Courtout F, Tollis S, Sagot I (2016) Quiescent Saccharomyces cerevisiae forms telomere hyperclusters at the nuclear membrane vicinity through a multifaceted mechanism involving Esc1, the Sir complex, and chromatin condensation. Mol Biol Cell 27:1875–1884
Lengronne A, Katou Y, Mori S, Yokobayashi S, Kelly GP, Itoh T, Watanabe Y, Shirahige K, Uhlmann F (2004) Cohesin relocation from sites of chromosomal loading to places of convergent transcription. Nature 430:573–578
Li L, Miles S, Breeden LL (2015) A genetic screen for saccharomyces cerevisiae mutants that fail to enter quiescence. G3 (Bethesda) 5:1783–1795
Litwin I, Wysocki R (2018) New insights into cohesin loading. Curr Genet 64:53–61
Lohr D, Ide G (1979) Comparison on the structure and transcriptional capability of growing phase and stationary yeast chromatin: a model for reversible gene activation. Nucleic Acids Res 6:1909–1927
Lupianez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161:1012–1025
Matityahu A, Onn I (2018) A new twist in the coil: functions of the coiled-coil domain of structural maintenance of chromosome (SMC) proteins. Curr Genet 64:109–116
Matsushima Y, Sakamoto N, Awazu A (2019) Insulator activities of nucleosome-excluding DNA sequences without bound chromatin looping proteins. J Phys Chem B 123:1035–1043
McKnight JN, Boerma JW, Breeden LL, Tsukiyama T (2015) Global promoter targeting of a conserved lysine deacetylase for transcriptional shutoff during quiescence entry. Mol Cell 59:732–743
Mizuguchi T, Fudenberg G, Mehta S, Belton JM, Taneja N, Folco HD, FitzGerald P, Dekker J, Mirny L, Barrowman J et al (2014) Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516:432–435
Ngubo M, Kemp G, Patterton HG (2011) Nano-electrospray tandem mass spectrometric analysis of the acetylation state of histones H3 and H4 in stationary phase in Saccharomyces cerevisiae. BMC Biochem 12:34
Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J et al (2012) Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485:381–385
Ocampo-Hafalla M, Munoz S, Samora CP, Uhlmann F (2016) Evidence for cohesin sliding along budding yeast chromosomes. Open Biol 6:15078
Paul MR, Markowitz TE, Hochwagen A, Ercan S (2018) Condensin depletion causes genome decompaction without altering the level of global gene expression in Saccharomyces cerevisiae. Genetics 210:331–344
Paul MR, Hochwagen A, Ercan S (2019) Condensin action and compaction. Curr Genet 65:407–415
Pinon R (1978) Folded chromosomes in non-cycling yeast cells: evidence for a characteristic g0 form. Chromosoma 67:263–274
Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159:1665–1680
Rao SSP, Huang SC, St Hilaire BG, Engreitz JM, Perez EM, Kieffer-Kwon KR, Sanborn AL, Johnstone SE, Bascom GD, Bochkov ID et al (2017) Cohesin loss eliminates all loop domains. Cell 171(305–320):e324
Rawlings JS, Gatzka M, Thomas PG, Ihle JN (2011) Chromatin condensation via the condensin II complex is required for peripheral T-cell quiescence. EMBO J 30:263–276
Robellet X, Vanoosthuyse V, Bernard P (2017) The loading of condensin in the context of chromatin. Curr Genet 63:577–589
Roche B, Arcangioli B, Martienssen R (2017) Transcriptional reprogramming in cellular quiescence. RNA Biol 14:843–853
Rutledge MT, Russo M, Belton JM, Dekker J, Broach JR (2015) The yeast genome undergoes significant topological reorganization in quiescence. Nucleic Acids Res 43:8299–8313
Sanborn AL, Rao SS, Huang SC, Durand NC, Huntley MH, Jewett AI, Bochkov ID, Chinnappan D, Cutkosky A, Li J et al (2015) Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc Natl Acad Sci USA 112:E6456–E6465
Sutani T, Sakata T, Nakato R, Masuda K, Ishibashi M, Yamashita D, Suzuki Y, Hirano T, Bando M, Shirahige K (2015) Condensin targets and reduces unwound DNA structures associated with transcription in mitotic chromosome condensation. Nat Commun 6:7815
Swygert SG, Kim S, Wu X, Fu T, Hsieh TH, Rando OJ, Eisenman RN, Shendure J, McKnight JN, Tsukiyama T (2019) Condensin-dependent chromatin compaction represses transcription globally during quiescence. Mol Cell 73(533–546):e534
Tanizawa H, Kim KD, Iwasaki O, Noma KI (2017) Architectural alterations of the fission yeast genome during the cell cycle. Nat Struct Mol Biol 24:965–976
Terakawa T, Bisht S, Eeftens JM, Dekker C, Haering CH, Greene EC (2017) The condensin complex is a mechanochemical motor that translocates along DNA. Science 358:672–676
Toselli-Mollereau E, Robellet X, Fauque L, Lemaire S, Schiklenk C, Klein C, Hocquet C, Legros P, N’Guyen L, Mouillard L et al (2016) Nucleosome eviction in mitosis assists condensin loading and chromosome condensation. EMBO J 35:1565–1581
Wang X, Hughes AC, Brandao HB, Walker B, Lierz C, Cochran JC, Oakley MG, Kruse AC, Rudner DZ (2018) In vivo evidence for ATPase-dependent DNA translocation by the Bacillus subtilis SMC condensin complex. Mol Cell 71(841–847):e845
Werner-Washburne M, Braun E, Johnston GC, Singer RA (1993) Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol Rev 57:383–401
Young CP, Hillyer C, Hokamp K, Fitzpatrick DJ, Konstantinov NK, Welty JS, Ness SA, Werner-Washburne M, Fleming AB, Osley MA (2017) Distinct histone methylation and transcription profiles are established during the development of cellular quiescence in yeast. BMC Genom 18:107
Yuen KC, Gerton JL (2018) Taking cohesin and condensin in context. PLoS Genet 14:e1007118
Yuen KC, Slaughter BD, Gerton JL (2017) Condensin II is anchored by TFIIIC and H3K4me3 in the mammalian genome and supports the expression of active dense gene clusters. Sci Adv 3:e1700191
Acknowledgements
S.G.S. has been supported by Grants F32GM120962 from NIGMS and T32CA009657 from NCI, and T.T. and S.G.S. were supported by NIGMS R01GM111428.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Swygert, S.G., Tsukiyama, T. Unraveling quiescence-specific repressive chromatin domains. Curr Genet 65, 1145–1151 (2019). https://doi.org/10.1007/s00294-019-00985-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-019-00985-9