
Abstract
Eukaryotic cells activate cell cycle checkpoints in response to DNA damage. In Saccharomyces cerevisiae, the DNA damage response is achieved by the activation of the sensor kinases Mec1 and Tel1 and transmitted to the effector kinase Rad53. Rad9 and Mrc1 are thought to differentially mediate the activation of Rad53 depending on the cell cycle phase. Rad9 can respond to DNA lesions throughout the cell cycle, whereas Mrc1 responds to replication impediments in S phase. It was not clear if Rad9 and Mrc1 were triggering the same response to DNA damage occurring in S phase. By carefully studying the kinetics of activation of Rad53 by different types of replication stresses, we recently showed that Rad9 and Mrc1 cooperate in time and space to trigger a unique response to DNA damage in S phase. This primarily includes the control of both DNA replication initiation and elongation. After showing that Rad9 plays a preponderant role during S phase, the data presented here provocatively suggest that Mrc1 could also mediate the activation of Rad53 outside of S phase.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Signaling pathways have evolved in eukaryotic cells in response to DNA damage to coordinate DNA repair with the progression of the cell cycle. These pathways activate checkpoints during any cell cycle phase to prevent the persistence, duplication or transmission of damaged DNA molecules. These actions allow the maintenance of genome integrity, thus safeguarding cellular functions. At the molecular level, these sophisticated signaling cascades involve protein kinases acting as sensors and effectors. These kinases phosphorylate various targets to achieve both the activation and transmission of the signal throughout the cell to trigger an adequate response to DNA damage (Ciccia and Elledge 2010). DNA damage is detected as discontinuities in the double-stranded DNA molecule, i.e., gaps or breaks. DNA gaps contain single-stranded DNA (ssDNA) and 5′ junctions between single-stranded and double-stranded DNA fragments. In the budding yeast Saccharomyces cerevisiae, these structures are, respectively, recognized by the heterotrimer RPA (Rfa1-Rfa2-Rfa3) and 9-1-1 (Ddc1-Rad17-Mec3) complexes (Majka and Burgers 2003; Rouse and Jackson 2002; Zou and Elledge 2003). On one hand, RPA allows the recruitment of the sensor kinase Mec1 through its binding partner Ddc2 and, on the other hand, 9-1-1 activates it (Majka et al. 2006; Navadgi-Patil and Burgers 2009; Paciotti et al. 2000). Mec1 can also be activated by the scaffold protein Dpb11 and the DNA helicase/nuclease Dna2 (Kumar and Burgers 2013; Navadgi-Patil and Burgers 2008). DNA double-strand breaks (DSBs) are recognized by the MRX complex (Mre11-Rad50-Xrs2) and the sensor kinase Tel1 (Grenon et al. 2001; Nakada et al. 2003). Repair of DSBs requires the processing of DSB ends, generating ssDNA tails that can recruit and activate Mec1. Mec1 then phosphorylates the effector kinase Rad53 (Ma et al. 2006; Sanchez et al. 1999, 1996). Yet, this step requires the involvement of mediator proteins that bring Rad53 in close proximity to Mec1 at the sites of DNA damage and promote its autophosphorylation.
Rad9 is one of such mediators (Weinert 1998). It is recruited to DNA damage sites through the interaction with modified histones H3, methylated on lysine 79 by Dot1, and H2A phosphorylated on serine 129 by Mec1/Tel1 (Downs et al. 2000; Giannattasio et al. 2005; Grenon et al. 2007; Lee et al. 2014; Toh et al. 2006; Wysocki et al. 2005). It can be stabilized there by interacting with Dpb11 and the 9-1-1 complex (di Cicco et al. 2017; Pfander and Diffley 2011). Rad9 becomes phosphorylated by Mec1 and is then able to recruit two Rad53 molecules to stimulate their autophosphorylation and activation (Emili 1998; Gilbert et al. 2001; Sweeney et al. 2005). Another mediator of Rad53 activation is Mrc1 (Alcasabas et al. 2001; Tanaka and Russell 2001). Mrc1 is a constitutive component of the replication fork, where it links the DNA polymerase ε with the replicative helicase component Mcm6 (Katou et al. 2003; Komata et al. 2009; Lou et al. 2008). It also forms a complex with Csm3 and Tof1 to promote normal fork progression (Bando et al. 2009; Calzada et al. 2005; Nedelcheva et al. 2005; Szyjka et al. 2005; Tourrière et al. 2005; Yeeles et al. 2017). As a consequence of its location, Mrc1-mediated activation of Rad53 is dependent on DNA replication during S phase. Any impediment to the progression of replication forks is thought to induce the uncoupling between the helicase and polymerase activities or between the leading and the lagging strand synthesis, which would lead to the accumulation of ssDNA and 5′ ssDNA/dsDNA junctions at unligated Okazaki fragments. Alternatively, fork stalling could induce the controlled degradation of nascent DNA, also leading to the accumulation of the same DNA structures. Both scenarios are suited to promote the recruitment and activation of Mec1, which phosphorylates Mrc1, in turn mediating the activation of Rad53 (Alcasabas et al. 2001; Chen and Zhou 2009; Osborn and Elledge 2003; Smolka et al. 2006; Tanaka and Russell 2004; Xu et al. 2006).
Opposite to this cell cycle phase-specific activation of Rad53 via Mrc1, Rad9 can virtually mediate Rad53 activation at any time (Siede et al. 1993; Weinert 1998). Nevertheless, the slowdown of S phase in response to damage in the template DNA or any other impediment to replication fork progression was described to be dependent on Mrc1 and not on Rad9 (Alcasabas et al. 2001; Katou et al. 2003). This has led to a conceptual separation of activating pathways of the DNA damage response (DDR) during S phase, Mrc1 triggering the DNA replication checkpoint (DRC), and Rad9 the DNA damage checkpoint (DDC) (Branzei and Foiani 2010; Crabbé et al. 2010; Pardo et al. 2017). Yet, irrespective of the way by which Mrc1 or Rad9 are invoked, each of them alone can mediate almost identical phosphorylation patterns of Rad53 (Chen et al. 2014). This fact questions the pertinence of defining two separate checkpoint pathways and prompted us to investigate the interplay between Rad9 and Mrc1 in response to DNA damage in S phase.
In our recent work by Bacal, Moriel-Carretero et al. (2018), we aimed at looking if the DDC could repress the activation of late-firing replication origins, a well-described function of Rad53 when activated by the DRC. As a matter of fact, replication fork stalling caused by the lack of dNTPs in cells treated with hydroxyurea (HU) induces the repression of late origins through Mrc1, not Rad9 (Crabbé et al. 2010). Unlike HU, MMS induces a Rad9-dependent activation of Rad53 (Emili 1998; Schwartz et al. 2002). However, Rad53 has been shown to repress late origins when cells replicate DNA in the presence of MMS (Tercero and Diffley 2001; Tercero et al. 2003), suggesting that both Rad9 and Mrc1 are required to orchestrate the DDR to MMS. Indeed, we observed that the repression of late origins in MMS-treated cells depends primarily on Mrc1, but we could see that Rad9 is also involved in this process if the amount of MMS-induced DNA lesions does not impede replication fork progression. In this situation, DNA lesions can be bypassed and left behind forks, where Rad9 can be recruited (Bacal et al. 2018). This is in complete agreement with a recent work by García-Rodríguez et al. (2018), which shows that Rad9-mediated activation of Rad53 (DDC) in response to MMS is triggered by ssDNA gaps that form on nascent DNA after re-priming of DNA synthesis downstream of the DNA lesions.
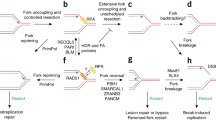
Nevertheless, the role of Rad9 in repressing late origins was minor in these conditions. We could propose an explanation for this minor contribution of Rad9 considering the kinetics of Rad53 activation mediated by Mrc1 and Rad9. Mrc1 is required for the fast but transient activation of Rad53, whereas Rad9 is necessary for a slower but sustained activation (Fig. 1). We think that this transient DRC signaling is linked to the physical association of Mrc1 to replication forks and to the observation that a threshold number of stalled forks needs to be reached to activate this pathway (Shimada et al. 2002; Tercero et al. 2003). As cells progress through S phase, adjacent replicons gradually merge and the number of active forks irremediably decreases, thus reducing the number of fork-associated Mrc1 molecules below the threshold required to activate the DRC. Consequently, replicated DNA regions accumulate and provide an increasing number of sites for Rad9 recruitment. The Rad9-mediated activation of Rad53 is slow because histones must be modified prior to Rad9 recruitment. Moreover, the ssDNA gaps left behind replication forks need to be processed for an efficient DDC activation (Galanti and Pfander 2018; García-Rodríguez et al. 2018). Finally, Rad53 activation by Rad9 is dampened by the Slx4–Rtt107 complex, which competes with Rad9 for the binding to DNA damage sites (Balint et al. 2015; Cussiol et al. 2015; Ohouo et al. 2013). In contrast, Mrc1 is an integral component of the replisome and is ideally positioned to mediate a fast activation of Rad53 in response to any replication impediment (Alcasabas et al. 2001). We reasoned that the DDC should therefore be able to efficiently repress the activation of late-firing origins if activated early enough. We have validated this hypothesis by activating the DDC with a limited amount of DNA double-strand breaks in cells blocked in early S phase and showed that Rad9 can efficiently mediate the repression of late origins when cells are allowed to resume replication (Bacal et al. 2018).

Schematic representation of the kinetics of Rad53 activation by the DRC and the DDC and the S phase checkpoint functions during replication stress. We propose that the transcriptional response to up-regulate the dNTP pools and to express the genes required for DNA repair (Chabes and Thelander 2003; de Bruin and Wittenberg 2009; Dmowski and Fijalkowska 2017; Mikolaskova et al. 2018) is triggered by the DRC as an early event. The repression of late origins (Yoshida et al. 2013) remains possible until the last origins have been fired, and is mainly a DRC function. Finally, the preservation of the fork ability to restart (Labib and De Piccoli 2011), the slow-down of fork progression and the cell cycle arrest before the end of mitosis (Palou et al. 2017) are functions that are shared by the DRC and DDC and that remain activated until the resolution of the replication stress
Additionally, our study unambiguously clarified that the DDR can downregulate the progression of replication forks when the template DNA is damaged. We first observed that rad9∆ mutants progressed faster through S phase than wild-type cells when exposed to MMS, and this could not be entirely explained by the modest derepression of a subset of late origins (Bacal et al. 2018). Nevertheless, this was not enough to claim that replication forks in wild type cells were being actively slowed down by the DDC signaling and not by the presence of an alkylated DNA template, even if we used a low dose of MMS. We then set up conditions to induce a limited number of DSBs with Zeocin (around 10 breaks per genome when cells enter into S phase) to induce the DDC without impeding replication initiation. This allowed us to follow the progression of individual replication forks under high DDC and low DRC activation. Our results showed that Rad9-mediated Rad53 activation at DSBs was slowing down replication progression in trans (Bacal et al. 2018) (Fig. 1). These results raised the question of whether the down-regulation of replication fork progression was specific to the DDC. We do not think this is the case because replication fork stalling is the trigger for DRC activation and this precludes to appreciate a fork slowing down mediated by Rad53. Conversely, we have reported earlier that the DRC and not the DDC could prevent homologous recombination (HR) events in S phase by inhibiting the end processing of DSBs (Alabert et al. 2009). Yet, the Rad9-dependent DDR to DSBs is a well-described negative regulator of DSB end resection (Bonetti et al. 2015; Ferrari et al. 2015). In light of our new results describing the kinetics of Rad53 activation by Mrc1 and Rad9, we can now propose that Rad9 could mediate the inhibition of HR by the DDR during S phase if the DDC were activated early enough. Altogether, we conclude that the DDC involving Rad9-mediated activation of Rad53 by Mec1 is the main response to DNA damage during all the phases of the cell cycle. As the regulation of S phase progression in response to DNA damage requires the timely activation of Rad53 to delay the activation of late-firing replication origins, cells have evolved a complementary pathway to activate Rad53 in early S phase (DRC) involving a specific component of the replisome, Mrc1. Rad9 and Mrc1 thus mediate the continuous activation of Rad53, cooperating to ensure the early and late functions of a unique response to DNA replication stress (Fig. 1).
Intriguingly, we noticed that Rad53 phosphorylation was not completely abolished in the rad9∆ mutant when DSBs were induced in G1 by Zeocin and cells released into S phase (Bacal et al. 2018) (Fig. 2). Indeed, we reproducibly observed a subtle phospho-shift of Rad53 from 90 min after release into S phase when cells had reached the G2 phase, as observed by flow cytometry (Fig. 2b, c). This was confirmed by the accumulation of cyclin B2 (Clb2), an indicator of cell entry into G2 phase (Fig. 2c). This was surprising, since the alternative pathway to activate Rad53 in the absence of Rad9 is only active in early S phase. Nevertheless, we have investigated whether this G2-specific phosphorylation of Rad53 depends on Mrc1 by repeating the experiment in the mrc1∆ rad9∆ double mutant. We show here that the absence of Mrc1 mildly but consistently decreased the phosphorylation of Rad53 (Fig. 2c, d), raising the possibility that Mrc1 could mediate the phosphorylation of Rad53 in late S or in G2 phases. As described by others (Doksani et al. 2009), replication forks encountering a DSB do not pause at the break site but are rapidly resolved into linear ends. This rules out the possibility that stalled forks accumulate at DSBs and reach the threshold required for DRC activation. Alternatively, this Mrc1-dependent signaling at persistent DSBs could relate to a DSB repair mechanism involving DNA synthesis such as Break-Induced Replication (BIR). BIR is described to occur during G2, in agreement with the kinetics we report. Yet, the DNA synthesis machinery involved in BIR differs from normal replication forks and does not contain MCM helicases nor DNA polymerase ε (Lydeard et al. 2007; Wilson et al. 2013). It is therefore unclear whether Mrc1 plays an active role in BIR and could mediate Rad53 activation during this process. Alternatively, Mrc1 could contribute to Rad53 activation independent of DNA synthesis, as reported earlier for its orthologue Claspin in Xenopus (Yoo et al. 2006).

Mrc1 can mediate Rad53 phosphorylation in late S/G2 in the absence of Rad9 in response to DSBs in S phase. a Wild-type (PP3372), mrc1Δ (PP1196), rad9Δ (PP1197) and mrc1Δ rad9Δ (PP1195) cells containing a 2 µm plasmid overexpressing RNR1 (Desany et al. 1998) were synchronized in G1 with α-factor. 100 µg/ml Zeocin was added to the medium and cells were released from the G1 arrest by the addition of pronase (75 µg/ml) 45 min later. Cells were collected at indicated time points. Protein extraction and Western blots were performed as described previously (Bacal et al. 2018), using anti-Rad53 (gift from C. Santocanale) and anti-Clb2 (y-180; SCBT) antibodies. b Analysis of DNA content by flow cytometry. c Analysis of Rad53 phosphorylation in G1 prior to Zeocin addition (G1) and at indicated times. d Densitometry profiles of Rad53 mobility shifts from c at 120 min. The increase in Rad53 phospho-shift is indicated by “P” and the arrow, from left to right
Still, low levels of Rad53 phosphorylation remained detectable in the mrc1∆ rad9∆ double mutant (Fig. 2c, d), suggesting that other factors mediate Rad53 activation in the absence of Mrc1 and Rad9. RFCCtf18 and Sgs1 have been shown to facilitate Mec1-dependent phosphorylation of Rad53 at replication forks stalled by dNTP depletion in HU-treated cells (Bjergbaek et al. 2005; Crabbé et al. 2010; Kubota et al. 2011). RFCCtf18 is an alternative loader/unloader for PCNA, a processivity factor for DNA polymerases (Bylund and Burgers 2005). It interacts with the DNA polymerase ε and has a function in sister-chromatid cohesion establishment during DNA replication (García-Rodríguez et al. 2015; Lengronne et al. 2006). Additionally, RFCCtf18 is absolutely required for the DRC activation, for which it is epistatic with Mrc1 (Crabbé et al. 2010). As RFCCtf18 has neither been described to be phosphorylated by Mec1/Tel1 in response to replication stress (Bastos de Oliveira et al. 2015; Chen et al. 2010; Smolka et al. 2007), nor to interact physically with Rad53, it cannot be considered as a canonical mediator of Rad53 activation. Instead, RFCCtf18 could be required to maintain the replisome in a conformation that allows Mec1 and Mrc1 to activate Rad53 in response to replication impediments. In response to DSBs, RFCCtf18 is also recruited to establish sister-chromatid cohesion but neither this process nor RFCCtf18 itself are required for Rad53 activation in response to DSBs (Crabbé et al. 2010; Ogiwara et al. 2007). All these data make RFCCtf18 a poor candidate for stimulating Rad53 phosphorylation in mrc1∆ rad9∆ cells in the presence of DSBs.
Unlike RFCCtf18, Sgs1 can interact directly with Rad53 after being phosphorylated by Mec1 (Hegnauer et al. 2012). Sgs1 is a DNA helicase that bears several functions in DSB processing, resolution of recombination intermediates and Rad53 activation (Bjergbaek et al. 2005; Cejka et al. 2010; Gravel et al. 2008; Mimitou and Symington 2008; Zhu et al. 2008). The absence of Sgs1 modestly affects the activation of Rad53 in cells exposed to HU or MMS and Sgs1 appears to act in the same pathway as Mrc1 for Rad53 activation (Bjergbaek et al. 2005; Hegnauer et al. 2012; Nielsen et al. 2013). The role of Sgs1 as a mediator of Rad53 activation is more evident in the absence of Rad9 or RFCRad24, another RFC-like complex responsible for the loading of the 9-1-1 complex (Bjergbaek et al. 2005; Hegnauer et al. 2012; Nielsen et al. 2013). For example, Rad53 activation in rad9∆ cells exposed to MMS is completely abolished in the absence of Sgs1 (Nielsen et al. 2013). Of importance, Rad53 is activated very late after S phase completion in rad9∆ cells treated with MMS (Nielsen et al. 2013), likely in G2/M, similarly to our observations in rad9∆ cells treated with Zeocin. MMS has been shown to induce the formation of chromatin bridges, which are structures connecting the sister chromatids during late mitosis (Germann et al. 2014). Such structures recruit Mec1 and Sgs1 and activate Rad53 (Germann et al. 2014). Thus, Sgs1 appears as a potential candidate for activating Rad53 in G2/M in the absence of Mrc1 and Rad9 in response to DNA damage. Further experiments will be needed to explore this possibility.
Finally, we do not discard the possibility that the sensor kinase Tel1 could be responsible for the residual Rad53 phosphorylation observed in mrc1∆ rad9∆ cells. In response to DSBs, Tel1 is able to phosphorylate Rad53 in the absence of Mec1, although less efficiently (Nakada et al. 2003). Supporting this hypothesis, Tel1 is involved in the resolution of replication termination at DSBs (Doksani et al. 2009).
Collectively, our recent work replaces Rad9 as a main actor of the DDR during S phase. The activation of the DDC by Rad9 allows Rad53 signaling to be maintained in time to ensure the integrity of challenged replication forks, slows down the progression of otherwise unchallenged forks and prevents the completion of mitosis until problems are solved. We also suggest that Mrc1 can unexpectedly signal DNA damage in late S or G2 (Fig. 2). Altogether, these data support a model in which the checkpoint response to DNA damage is a single pathway with the sufficient versatility as to integrate temporal and spatial cues to safeguard the genome integrity during the cell cycle.
References
Alabert C, Bianco JN, Pasero P (2009) Differential regulation of homologous recombination at DNA breaks and replication forks by the Mrc1 branch of the S-phase checkpoint. EMBO J 28:1131–1141
Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ (2001) Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 3:958–965
Bacal J, Moriel Carretero M, Pardo B, Barthe A, Sharma S, Chabes A, Lengronne A, Pasero P (2018) Mrc1 and Rad9 cooperate to regulate initiation and elongation of DNA replication in response to DNA damage. EMBO J. https://doi.org/10.15252/embj.201899319
Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, Yimit A, Ou J, Nakato R, Gurevich A, Shirahige K, Smolka MB, Zhang Z, Brown GW (2015) Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J 34:2182–2197
Bando M, Katou Y, Komata M, Tanaka H, Itoh T, Sutani T, Shirahige K (2009) Csm3, Tof1, and Mrc1 form a heterotrimeric mediator complex that associates with DNA replication forks. J Biol Chem 284:34355–34365
Bastos de Oliveira FM, Kim D, Cussiol José R, Das J, Jeong Min C, Doerfler L, Schmidt Kristina H, Yu H, Smolka Marcus B (2015) Phosphoproteomics reveals distinct modes of Mec1/ATR signaling during DNA replication. Mol Cell 57:1124–1132
Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM (2005) Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J 24:405–417
Bonetti D, Villa M, Gobbini E, Cassani C, Tedeschi G, Longhese MP (2015) Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep 16:351–361
Branzei D, Foiani M (2010) Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11:208–219
Bylund GO, Burgers PMJ (2005) Replication protein A-directed unloading of PCNA by the Ctf18 cohesion establishment complex. Mol Cell Biol 25:5445–5455
Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K (2005) Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev 19:1905–1919
Cejka P, Cannavo E, Polaczek P, Masuda-Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC (2010) DNA end resection by Dna2-Sgs1-RPA and its stimulation by Top3-Rmi1 and Mre11-Rad50-Xrs2. Nature 467:112–116
Chabes A, Thelander L (2003) DNA building blocks at the foundation of better survival. Cell Cycle 2:171–173
Chen S-h, Zhou H (2009) Reconstitution of Rad53 activation by Mec1 through adaptor protein Mrc1. J Biol Chem 284:18593–18604
Chen S-h, Albuquerque CP, Liang J, Suhandynata RT, Zhou H (2010) A proteome-wide analysis of kinase-substrate network in the DNA damage response. J Biol Chem 285:12803–12812
Chen ES, Hoch NC, Wang SC, Pellicioli A, Heierhorst J, Tsai MD (2014) Use of quantitative mass spectrometric analysis to elucidate the mechanisms of phospho-priming and auto-activation of the checkpoint kinase Rad53 in vivo. Mol Cell Proteom MCP 13:551–565
Ciccia A, Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40:179–204
Crabbé L, Thomas A, Pantesco V, De Vos J, Pasero P, Lengronne A (2010) Analysis of replication profiles reveals key role of RFC-Ctf18 in yeast replication stress response. Nat Struct Mol Biol 17:1391–1397
Cussiol JR, Jablonowski CM, Yimit A, Brown GW, Smolka MB (2015) Dampening DNA damage checkpoint signalling via coordinated BRCT domain interactions. EMBO J 34:1704–1717
de Bruin RA, Wittenberg C (2009) All eukaryotes: before turning off G1-S transcription, please check your DNA. Cell Cycle 8:214–217
Desany BA, Alcasabas AA, Bachant JB, Elledge SJ (1998) Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev 12:2956–2970
di Cicco G, Bantele SCS, Reusswig K-U, Pfander B (2017) A cell cycle-independent mode of the Rad9-Dpb11 interaction is induced by DNA damage. Sci Rep 7:11650–11650
Dmowski M, Fijalkowska IJ (2017) Diverse roles of Dpb2, the non-catalytic subunit of DNA polymerase epsilon. Curr Genet 63:983–987
Doksani Y, Bermejo R, Fiorani S, Haber JE, Foiani M (2009) Replicon dynamics, dormant origin firing, and terminal fork integrity after double-strand break formation. Cell 137:247–258
Downs JA, Lowndes NF, Jackson SP (2000) A role for Saccharomyces cerevisiae histone H2A in DNA repair. Nature 408:1001–1004
Emili A (1998) MEC1-dependent phosphorylation of Rad9p in response to DNA damage. Mol Cell 2:183–189
Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, Lazzaro F, Tsabar M, Marini F, Haber JE, Pellicioli A (2015) Functional interplay between the 53BP1-ortholog Rad9 and the Mre11 complex regulates resection, end-tethering and repair of a double-strand break. PLoS Genet 11:e1004928
Galanti L, Pfander B (2018) Right time, right place-DNA damage and DNA replication checkpoints collectively safeguard S phase. EMBO J. https://doi.org/10.15252/embj.2018100681
García-Rodríguez LJ, De Piccoli G, Marchesi V, Jones RC, Edmondson RD, Labib K (2015) A conserved Polϵ binding module in Ctf18-RFC is required for S-phase checkpoint activation downstream of Mec1. Nucleic Acids Res 43:8830–8838
García-Rodríguez N, Morawska M, Wong RP, Daigaku Y, Ulrich HD (2018) Spatial separation between replisome- and template-induced replication stress signaling. EMBO J. https://doi.org/10.15252/embj.201798369
Germann SM, Schramke V, Pedersen RT, Gallina I, Eckert-Boulet N, Oestergaard VH, Lisby M (2014) TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J Cell Biol 204:45–59
Giannattasio M, Lazzaro F, Plevani P, Muzi-Falconi M (2005) The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6-Bre1 and H3 methylation by Dot1. J Biol Chem 280:9879–9886
Gilbert CS, Green CM, Lowndes NF (2001) Budding yeast Rad9 is an ATP-dependent Rad53 activating machine. Mol Cell 8:129–136
Gravel S, Chapman JR, Magill C, Jackson SP (2008) DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev 22:2767–2772
Grenon M, Gilbert C, Lowndes NF (2001) Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat Cell Biol 3:844–847
Grenon M, Costelloe T, Jimeno S, O’Shaughnessy A, Fitzgerald J, Zgheib O, Degerth L, Lowndes NF (2007) Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast 24:105–119
Hegnauer AM, Hustedt N, Shimada K, Pike BL, Vogel M, Amsler P, Rubin SM, van Leeuwen F, Guenole A, van Attikum H, Thoma NH, Gasser SM (2012) An N-terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J 31:3768–3783
Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K (2003) S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature 424:1078–1083
Komata M, Bando M, Araki H, Shirahige K (2009) The direct binding of Mrc1, a checkpoint mediator, to Mcm6, a replication helicase, is essential for the replication checkpoint against methyl methanesulfonate-induced stress. Mol Cell Biol 29:5008–5019
Kubota T, Hiraga S-i, Yamada K, Lamond AI, Donaldson AD (2011) Quantitative proteomic analysis of chromatin reveals that Ctf18 acts in the DNA replication checkpoint. Mol Cell Proteom 10:M110.005561
Kumar S, Burgers PM (2013) Lagging strand maturation factor Dna2 is a component of the replication checkpoint initiation machinery. Genes Dev 27:313–321
Labib K, De Piccoli G (2011) Surviving chromosome replication: the many roles of the S-phase checkpoint pathway. Philos Trans R Soc Lond Ser B Biol Sci 366:3554–3561
Lee CS, Lee K, Legube G, Haber JE (2014) Dynamics of yeast histone H2A and H2B phosphorylation in response to a double-strand break. Nat Struct Mol Biol 21:103–109
Lengronne A, McIntyre J, Katou Y, Kanoh Y, Hopfner KP, Shirahige K, Uhlmann F (2006) Establishment of sister chromatid cohesion at the S. cerevisiae replication fork. Mol Cell 23:787–799
Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL (2008) Mrc1 and DNA polymerase epsilon function together in linking DNA replication and the S phase checkpoint. Mol Cell 32:106–117
Lydeard JR, Jain S, Yamaguchi M, Haber JE (2007) Break-induced replication and telomerase-independent telomere maintenance require Pol32. Nature 448:820–823
Ma J-L, Lee S-J, Duong JK, Stern DF (2006) Activation of the checkpoint kinase Rad53 by the phosphatidyl inositol kinase-like kinase Mec1. J Biol Chem 281:3954–3963
Majka J, Burgers PM (2003) Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc Natl Acad Sci USA 100:2249–2254
Majka J, Niedziela-Majka A, Burgers PM (2006) The checkpoint clamp activates Mec1 kinase during initiation of the DNA damage checkpoint. Mol Cell 24:891–901
Mikolaskova B, Jurcik M, Cipakova I, Kretova M, Chovanec M, Cipak L (2018) Maintenance of genome stability: the unifying role of interconnections between the DNA damage response and RNA-processing pathways. Curr Genet 64:971–983
Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature 455:770–774
Nakada D, Matsumoto K, Sugimoto K (2003) ATM-related Tel1 associates with double-strand breaks through an Xrs2-dependent mechanism. Genes Dev 17:1957–1962
Navadgi-Patil VM, Burgers PM (2008) Yeast DNA replication protein Dpb11 activates the Mec1/ATR checkpoint kinase. J Biol Chem 283:35853–35859
Navadgi-Patil VM, Burgers PM (2009) A tale of two tails: Activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair 8:996–1003
Nedelcheva MN, Roguev A, Dolapchiev LB, Shevchenko A, Taskov HB, Stewart AF, Stoynov SS (2005) Uncoupling of unwinding from DNA synthesis implies regulation of MCM helicase by Tof1/Mrc1/Csm3 checkpoint complex. J Mol Biol 347:509–521
Nielsen I, Bentsen IB, Andersen AH, Gasser SM, Bjergbaek L (2013) A Rad53 independent function of Rad9 becomes crucial for genome maintenance in the absence of the RecQ Helicase Sgs1. PloS One 8:e81015
Ogiwara H, Ohuchi T, Ui A, Tada S, Enomoto T, Seki M (2007) Ctf18 is required for homologous recombination-mediated double-strand break repair. Nucl Acids Res 35:4989–5000
Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493:120–124
Osborn AJ, Elledge SJ (2003) Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 17:1755–1767
Paciotti V, Clerici M, Lucchini G, Longhese MP (2000) The checkpoint protein Ddc2, functionally related to S. pombe Rad26, interacts with Mec1 and is regulated by Mec1-dependent phosphorylation in budding yeast. Genes Dev 14:2046–2059
Palou R, Palou G, Quintana DG (2017) A role for the spindle assembly checkpoint in the DNA damage response. Curr Genet 63:275–280
Pardo B, Crabbé L, Pasero P (2017) Signaling pathways of replication stress in yeast. FEMS Yeast Res. https://doi.org/10.1093/femsyr/fow101
Pfander B, Diffley JF (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle-regulated Rad9 recruitment. EMBO J 30:4897–4907
Rouse J, Jackson SP (2002) Interfaces between the detection, signaling, and repair of DNA damage. Science 297:547–551
Sanchez Y, Desany BA, Jones WJ, Liu Q, Wang B, Elledge SJ (1996) Regulation of RAD53 by the ATM-like kinases MEC1 and TEL1 in yeast cell cycle checkpoint pathways. Science 271:357–360
Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ (1999) Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science 286:1166–1171
Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF (2002) Rad9 phosphorylation sites couple Rad53 to the Saccharomyces cerevisiae DNA damage checkpoint. Mol Cell 9:1055–1065
Shimada K, Pasero P, Gasser SM (2002) ORC and the intra-S-phase checkpoint: a threshold regulates Rad53p activation in S phase. Genes Dev 16:3236–3252
Siede W, Friedberg AS, Friedberg EC (1993) RAD9-dependent G1 arrest defines a second checkpoint for damaged DNA in the cell cycle of Saccharomyces cerevisiae. Proc Natl Acad Sci USA 90:7985–7989
Smolka MB, Chen S-h, Maddox PS, Enserink JM, Albuquerque CP, Wei XX, Desai A, Kolodner RD, Zhou H (2006) An FHA domain-mediated protein interaction network of Rad53 reveals its role in polarized cell growth. J Cell Biol 175:743–753
Smolka MB, Albuquerque CP, Chen SH, Zhou H (2007) Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA 104:10364–10369 doi
Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D (2005) Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol CB 15:1364–1375
Szyjka SJ, Viggiani CJ, Aparicio OM (2005) Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol Cell 19:691–697 doi
Tanaka K, Russell P (2001) Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat Cell Biol 3:966–972
Tanaka K, Russell P (2004) Cds1 phosphorylation by Rad3-Rad26 kinase is mediated by forkhead-associated domain interaction with Mrc1. J Biol Chem 279:32079–32086
Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412:553–557
Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11:1323–1336
Toh GWL, O’Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O’Rorke A, Lowndes NF (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5:693–703
Tourrière H, Versini G, Cordón-Preciado V, Alabert C, Pasero P (2005) Mrc1 and Tof1 promote replication fork progression and recovery independently of Rad53. Mol Cell 19:699–706
Weinert T (1998) DNA damage checkpoints update: getting molecular. Curr Opin Genet Dev 8:185–193
Wilson MA, Kwon Y, Xu Y, Chung WH, Chi P, Niu H, Mayle R, Chen X, Malkova A, Sung P, Ira G (2013) Pif1 helicase and Poldelta promote recombination-coupled DNA synthesis via bubble migration. Nature 502:393–396
Wysocki R, Javaheri A, Allard S, Sha F, Cote J, Kron SJ (2005) Role of Dot1-dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol 25:8430–8443
Xu Y, Davenport M, Kelly TJ (2006) Two-stage mechanism for activation of the DNA replication checkpoint kinase Cds1 in fission yeast. Genes Dev 20:990–1003
Yeeles JTP, Janska A, Early A, Diffley JFX (2017) How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell 65:105–116
Yoo HY, Jeong S-Y, Dunphy WG (2006) Site-specific phosphorylation of a checkpoint mediator protein controls its responses to different DNA structures. Genes Dev 20:772–783
Yoshida K, Poveda A, Pasero P (2013) Time to be versatile: regulation of the replication timing program in budding yeast. J Mol Biol 425:4696–4705
Zhu Z, Chung W-H, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell 134:981–994
Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP Recognition of RPA–ssDNA complexes. Science 300:1542–1548
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Moriel-Carretero, M., Pasero, P. & Pardo, B. DDR Inc., one business, two associates. Curr Genet 65, 445–451 (2019). https://doi.org/10.1007/s00294-018-0908-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-018-0908-7