
Abstract
Progression through the cell cycle is driven by the activities of the cyclin-dependent kinase (CDK) family of enzymes, which establish an ordered passage through the cell cycle phases. CDK activity is crucial for the cellular transitions from G1 to S and G2 to M, which are highly controlled to promote the faithful duplication of the genetic material and the transmission of the genome into daughter cells, respectively. While oscillations in CDK activity are essential for cell division, how its specific dynamics may shape cellular processes remains an open question. Recently, we have investigated the potential role of CDK in establishing the profile of replication initiation along the chromosomes, also referred to as the replication program. Our results demonstrated that the timing and level of CDK activity at G1/S provide two critical and independent inputs that modulate the pattern of origin usage. In this review, we will present the conclusions of our study and discuss the implications of our findings for cellular function and physiology.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cell proliferation requires a series of events that occurs in a given order to ensure that the genetic material is properly copied and propagated. Cell cycle progression is brought about by oscillations in the activities of the cyclin-dependent kinase (CDK) family of enzymes, which phosphorylate a variety of substrates during the cycle (Morgan 2007). The entry of cells into a new cycle is modulated by growth and nutritional signals, and it begins with the passage through a point in G1 in which cells become committed to genome duplication [START in yeast, the restriction point (R) in mammalian cells]. The ensuing multistep process of replication initiation is tightly controlled to reproduce the genome once and only once per cell cycle, and this relies on CDK function (Siddiqui et al. 2013). Abnormal increases in CDK activity, for instance through overexpression of cyclin partners or removal of CDK inhibitors, shorten G1 and have deleterious consequences for genome duplication and maintenance (Lengronne and Schwob 2002; Tanaka and Diffley 2002; Ekholm-Reed et al. 2004). Thus, CDKs are essential not only for triggering S phase entry but also for promoting genome integrity.
Interestingly, it has become increasingly clear that the complex mechanisms that govern DNA replication establish temporal and spatial patterns of replication origin usage along the chromosomes in diverse model systems. Replication programs are conserved between related species (Ryba et al. 2010; Yaffe et al. 2010; Muller and Nieduszynski 2012), suggesting a biological importance for the organization of genome duplication. These profiles have also been demonstrated to be sensitive to external conditions and to cellular states. Indeed, nitrogen availability modulates the profile of replication origin usage in the fission yeast (Wu and Nurse 2014), and the differentiation of mouse and human embryonic stem cells is coupled with alterations in replication patterns (Hiratani et al. 2008; Desprat et al. 2009; Pope et al. 2010). In a pathological context, changes in replication timing have been associated with different diseases, including cancers (Donley and Thayer 2013). Importantly, there is accumulating evidence that the program of DNA replication makes key contributions to cellular function. For example, replication timing has been shown to be involved in the control of gene expression (Müller and Nieduszynski 2017), and we have previously found that origin selection delineates the distribution of the double-stranded DNA breaks that are required for meiotic recombination (Wu and Nurse 2014). Nevertheless, we are only beginning to understand the mechanisms that determine this program (Wu and Nurse 2009; Aparicio 2013; Masai et al. 2017; Fu et al. 2018). Our recent studies have uncovered critical roles for the temporal and quantitative regulation of CDK activity at G1/S in modulating the organization of DNA replication (Perrot et al. 2018). As CDK function is central to a variety of cellular and developmental processes as well as to the response to environmental cues, our findings provide new perspectives into the interactions between cell cycle regulation, genome duplication, and cellular physiology.
Qualitative and quantitative aspects of CDK function in DNA replication
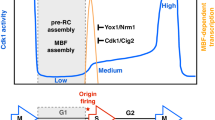
While CDKs bind with different cyclin partners for their activation, a number of studies have shown that cyclin/CDK diversity is not essential for cell proliferation. A high level of redundancy in cyclin-CDK functions has been demonstrated from yeast to mammals. For example, mouse embryonic fibroblasts lacking all three D-type cyclins that normally function in early G1 phase are able to proliferate (Kozar et al. 2004), and Cdk1 itself supports cell cycle events in the absence of all interphase CDKs in the early mouse embryo (Santamaría et al. 2007). In the fission yeast Schizosaccharomyces pombe, which harbors four cell cycle cyclins, the division cycle can be sustained by a single cyclin B (Cdc13) together with the sole cell cycle CDK (Cdc2) (Fisher and Nurse 1996; Coudreuse and Nurse 2010). Remarkably, imposed oscillations in the activity of a Cdc13–Cdc2 fusion protein are sufficient to trigger passage through the cell cycle phases, even in the absence of all other cell cycle cyclins (Coudreuse and Nurse 2010). These studies thus indicate that cell cycle transitions simply rely on attaining particular thresholds of CDK activity (low for S, high for M) and that oscillation of this activity promotes the ordered progression of cell cycle events.
In the context of replication initiation, CDK activity is a central regulator of two key steps: the assembly of the pre-replicative complex (pre-RC), which licenses potential origins, and the subsequent formation of the pre-initiation complex (pre-IC), which activates origins for DNA synthesis (Araki 2010; Siddiqui et al. 2013). In metazoa, multiple CDKs and their cyclin partners are involved in driving the cell cycle and promoting distinct phase transitions, with Cdks 1 and 2 as well as cyclins A and E playing roles during G1 and S (Morgan 2007; Harashima et al. 2013). Less complex organisms such as the budding yeast Saccharomyces cerevisiae and the fission yeast Schizosaccharomyces pombe likewise possess multiple G1 and S phase cyclins, although they have only one cell cycle CDK (Morgan 2007). Interestingly, different cyclin-CDK combinations have been suggested to have specific functions in genome duplication. Indeed, full replication efficacy in Xenopus egg extracts requires both endogenous cyclin E-Cdk2 and cyclin A-Cdk1 pairs (Krasinska et al. 2008), and specific cyclin-CDK complexes may regulate distinct origin subsets in the budding yeast (Donaldson et al. 1998). However, the importance of cyclin-CDK diversity for DNA replication has been challenged by multiple studies. For instance, budding yeast mutants lacking the S phase cyclins undergo a delayed S phase that activates both early and late firing replication origins (Donaldson et al. 1998), and the Clb2 mitotic cyclin is also able to perform S phase functions (Hu and Aparicio 2005). In addition, in Xenopus egg extracts, the decrease in DNA synthesis resulting from depletion of particular CDKs can be complemented by heterologous CDK complexes (Krasinska et al. 2008). Altogether, these findings support a quantitative requirement for CDK activity in S phase entry, revealing conflicting views of how CDK regulates genome duplication.
CDK timing and levels determine the organization of DNA replication
Despite our extensive knowledge of CDK regulators and targets (Bertoli et al. 2013; Bell and Labib 2016; Swaffer et al. 2016; Touati et al. 2018), it remained unknown whether establishing the genome-wide profile of origin usage requires both qualitative and quantitative components of CDK function. This has been difficult to investigate in vivo due in part to the presence of multiple cyclin-CDK complexes. In particular, it was not possible to dissociate potential qualitative differences in substrate phosphorylation provided by different cyclin-CDK pairs from quantitative changes in the dynamics and levels of overall CDK activity. Recently, we have addressed these fundamental questions and investigated the role of CDK in regulating the replication program (Perrot et al. 2018). To this end, we took advantage of a unique system in the fission yeast S. pombe that replaces the endogenous cell cycle circuit with a simplified CDK module whose activity can be precisely modulated via chemical genetics (Coudreuse and Nurse 2010). In these cells, the G1 and S phase cell cycle cyclins (Cig1, Cig2, and Puc1) are absent, and a fusion protein consisting of the mitotic cyclin (Cdc13) and the Cdk1 (Cdc2) autonomously drives cell proliferation with no detectable phenotypes (Coudreuse and Nurse 2010). This simplified system is not subject to many of the constraints of the endogenous network, which include the regulation of cyclin-CDK binding, the specific expression and degradation of distinct cyclins, and the differential localization of cyclins and CDKs (Morgan 2007). Moreover, a modification of the kinase moiety of the Cdc13–Cdc2 fusion protein allows for reversible and dose-dependent inhibition of its kinase activity by non-hydrolyzable ATP analogs (Bishop et al. 2000), enabling us to alter a single CDK activity with high temporal and quantitative resolution. This module, therefore, represents a powerful tool for dissecting the role of CDK activity in establishing the replication program.
Using this approach, we began by demonstrating that different cyclin-CDK complexes are not required to regulate specific groups of origins. Our results showed that a single qualitative CDK activity is sufficient to establish regional domains of origin timing and efficiency that are virtually identical to those in wild-type cells (Fig. 1). We then independently targeted two critical features of CDK activity: the timing at which cells accumulate sufficient activity for S phase entry and the level of CDK activity at S phase onset. First, we found that prolonging G1 through CDK inhibition induced alterations in pre-IC formation that led to an equalization of origin usage between replication domains (Fig. 2a). Origin activity was increased in regions of low efficiency, while in efficient domains this was unchanged or modestly reduced. This signature change occurred even with short extensions of G1 phase, with longer G1 durations inducing progressively greater changes. This demonstrates that the replication program is very sensitive to delays in CDK function. Next, we determined the quantitative effect of CDK activity on the pattern of replication initiation. Our system enabled us to induce cells to undergo S phase with a range of CDK activities while maintaining the same G1 length, thus uncoupling the timing of CDK function from its activity level (Fig. 2b). Our results showed that overall origin efficiencies display a dose-dependent response to the level of CDK activity at G1/S. In contrast to what we observed for G1 extensions, all chromosomal regions responded similarly to the changes in CDK activity levels, implying that this activity at the start of S phase is a direct and quantitative regulator of origin usage genome-wide. Collectively, our findings identify independent inputs for the temporal and quantitative regulation of CDK function in the organization of genome duplication.

Cyclin-CDK diversity is not required for establishing a normal replication program. Top left: diagram of the cell cycle cyclins (Cig1, Puc1, Cig2, and Cdc13) and CDK (Cdc2) that are present in wild-type fission yeast cells. Bottom left: diagram of the Cdc13–Cdc2 fusion protein that is sufficient to drive the cell cycle in the absence of all other cell cycle cyclins (Coudreuse and Nurse 2010). Right: representation of the replication program in wild-type (top) and Cdc13–Cdc2 (bottom). x-axis: chromosome coordinates, y-axis: origin activity. Origin activity (dashed line) represents the efficiency of origin usage, which is measured by the frequency of initiation at a given origin in a population of cells. Note that there is a correlation between the timing and efficiency of origin firing: early firing origins tend to be efficient (Heichinger et al. 2006). The wild-type and Cdc13–Cdc2 genetic backgrounds produce virtually identical profiles of replication initiation (Perrot et al. 2018)

Temporal and quantitative regulation of the replication program by CDK. a G1 extension due to CDK inhibition results in an equalization of replication timing and efficiency domains. Left: schematic of CDK activity patterns in cells with a normal (black) or prolonged (dashed blue) G1. TM: mitotic threshold, TS: S phase threshold. x-axis: cell cycle progression, y-axis: CDK activity. Right: representation of the replication programs (dashed line) in cells with a normal (top) or prolonged (bottom) G1. x-axis: chromosome coordinates, y-axis: origin activity. Upon G1 extension, inefficient domains show increased origin activity, while efficient regions are unaffected or show a modest decrease in origin usage (Perrot et al. 2018). b CDK activity is a quantitative regulator of genome-wide origin efficiencies. Left: schematic of CDK activity levels in cells undergoing S phase with different concentrations of the specific inhibitor. Lower CDK activities are associated with higher inhibitor concentrations (indicated by the different colors). G1 length is constant in these experiments. TM: mitotic threshold, TS: S phase threshold. x-axis: cell cycle progression, y-axis: CDK activity. Right: representation of the replication patterns (dashed lines) in the conditions depicted in the left panel. Colors are as in the left panel. x-axis: chromosome coordinates, y-axis: origin activity. Overall origin usage responds in a dose-dependent manner to the CDK activity level at S phase onset (Perrot et al. 2018)
The use of a simplified cyclin-CDK system has allowed us to reveal key principles underlying the regulation of DNA replication by CDK that may extend to cells containing the full complement of cyclins and CDKs. Multiple cyclin-CDK pairs have been described in all eukaryotic systems studied to date, and these complexes, which have distinct expression, degradation and activation patterns (Morgan 1997), may permit both flexibility and a fine level of control of overall CDK function. For instance, specific pairs may respond to different signals, and the combination of each of these activities may generate complex global activity profiles. The ensuing alterations in the organization of genome duplication may then contribute to the cellular responses to internal or external stimuli.
CDK function may coordinate replication with other cellular processes
Our results demonstrate that CDK activity is not only essential for triggering the onset of DNA replication but also participates in an additional layer of control that governs the profile of origin usage across the genome. Given that CDK activity is involved in numerous cellular pathways, it is tempting to speculate about the potential biological impact of this regulation. During the cell cycle, progression through the different phases is associated with specific waves of gene expression (Rustici et al. 2004; Bähler 2005; Hendler et al. 2018), and CDK activity has been identified as a direct regulator of this periodic transcription (Banyai et al. 2016; Rahi et al. 2016). The capacity of CDK to concomitantly regulate transcription and replication makes it a particularly appropriate input for coordinating these processes during G1/S. This idea is consistent with a recent study investigating the overexpression of cyclin E, an activator of Cdk2 in human cells that functions in S phase regulation. Deregulation of cyclin E is found in a variety of cancers, and this is associated with replication defects and genome instability (Spruck et al. 1999; Ekholm-Reed et al. 2004; Teixeira et al. 2015). Upon cyclin E overexpression, G1 is shortened and cells enter S phase prematurely, with DNA synthesis initiating from intragenic origins that are normally suppressed by transcription through these sites in G1 (Macheret and Halazonetis 2018). This then generates conflicts between these initiation events and the transcription machinery that lead to replication fork collapse and genome instability. Thus, an important function of CDK in G1/S may be to regulate both transcription and replication to preserve genome integrity.
In addition, a number of studies have identified a positive interplay between replication and transcription. For instance, early replication domains are correlated with transcriptional activity, and changes in replication timing during differentiation are associated with alterations in chromosome architecture and gene expression in mammalian cells (Rivera-Mulia et al. 2015, 2018). A link between replication initiation and transcription in C. elegans embryonic development has also been suggested (Pourkarimi et al. 2016; Rodríguez-Martínez et al. 2017). Notably, direct evidence for the importance of replication timing in gene expression was provided by work investigating the histone genes, whose transcription is tightly regulated to ensure packaging of newly synthesized DNA into chromatin (Hereford et al. 1981). Indeed, the early replication of histone loci is conserved in divergent budding yeast species, and delaying the duplication of HTA1-HTB1, one of the loci that encodes for histones H2A and H2B in S. cerevisiae, resulted in a reduction in its expression (Müller and Nieduszynski 2017). These findings indicate that the control of replication timing by CDK may contribute to establishing distinct transcriptional programs during key cellular transitions.
CDK regulation of the replication program may also be also central to other aspects of cellular physiology. We have investigated this in the context of meiosis, when DNA synthesis is followed by meiotic recombination and two rounds of chromosome segregation. The coupling of replication and recombination during meiosis has long been observed (Borde et al. 2000), but the mechanisms by which this occurs remained elusive. We demonstrated that the profile of origin usage along the chromosomes modulates the programmed formation of double-stranded DNA breaks for meiotic recombination (Wu and Nurse 2014). This and other studies (Murakami and Keeney 2014) therefore implicate the temporal and spatial pattern of DNA replication as a modulator of genetic diversity during this specialized cell cycle.
Taken together, one attractive idea that emerges is that the organization of DNA replication makes integral contributions to cellular transitions, and CDK function would be a versatile mechanism by which this regulation is achieved.
Perspectives for cell proliferation and cell fate
G1 is a critical period during which cells prepare for genome duplication, but it is also a time when cells receive signals that promote continued self-renewal, differentiation, or cell cycle exit (Dalton 2015). The length of G1 is flexible and is modulated by many parameters, including the availability of growth factors and the nutritional state of the cells. In the “extreme” case, G1 is not present, as seen in initial divisions of embryonic development in some organisms, which are comprised of only S and M phases (Foe and Alberts 1983; Farrell and O’Farrell 2014). G1 is short in natural populations of stem cells, and a prolongation of G1 is associated with differentiation (Lange and Calegari 2010; Coronado et al. 2013; Homem et al. 2015; Boward et al. 2016). Interestingly, increasing G1 length is necessary and sufficient to induce the differentiation of neural progenitors (Lange et al. 2009), and this parameter has been proposed to play a role in modulating the behaviors of distinct stem cells (Lange and Calegari 2010; Boward et al. 2016). Our work suggests that G1 duration may participate in these processes through changing replication organization, with the level of CDK activity at G1/S providing an additional layer of regulation. Consistent with this, different replication patterns are observed between embryonic stages and somatic cells in Xenopus (Laskey 1985; Hyrien et al. 1995; Walter and Newport 1997) as well as between mammalian stem cells and differentiated cells (Hiratani et al. 2008, 2010; Wilson et al. 2016). While it remains to be determined whether these alterations promote the accompanying physiological transitions, these findings collectively highlight intriguing links between the cellular state, the length of G1, and the organization of DNA replication. Given the crucial functions of CDKs in cell proliferation and fate decisions, our discovery of the role of CDK in establishing the replication program leads to exciting perspectives for understanding how genome duplication is coordinated with and contributes to the changes that occur during cellular adaptation and development.
References
Aparicio OM (2013) Location, location, location: it’s all in the timing for replication origins. Genes Dev 27:117–128. https://doi.org/10.1101/gad.209999.112
Araki H (2010) Cyclin-dependent kinase-dependent initiation of chromosomal DNA replication. Curr Opin Cell Biol 22:766–771. https://doi.org/10.1016/j.ceb.2010.07.015
Bähler J (2005) Cell-cycle control of gene expression in budding and fission yeast. Annu Rev Genet 39:69–94. https://doi.org/10.1146/annurev.genet.39.110304.095808
Banyai G, Baïdi F, Coudreuse D, Szilagyi Z (2016) Cdk1 activity acts as a quantitative platform for coordinating cell cycle progression with periodic transcription. Nat Commun 7:11161. https://doi.org/10.1038/ncomms11161
Bell SP, Labib K (2016) Chromosome duplication in Saccharomyces cerevisiae. Genetics 203:1027–1067. https://doi.org/10.1534/genetics.115.186452
Bertoli C, Skotheim JM, de Bruin RAM (2013) Control of cell cycle transcription during G1 and S phases. Nat Publ Group 14:518–528. https://doi.org/10.1038/nrm3629
Bishop AC, Ubersax JA, Petsch DT et al (2000) A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407:395–401. https://doi.org/10.1038/35030148
Borde V, Goldman AS, Lichten M (2000) Direct coupling between meiotic DNA replication and recombination initiation. Science 290:806–809
Boward B, Wu T, Dalton S (2016) Concise review: control of cell fate through cell cycle and pluripotency networks. Stem Cells 34:1427–1436. https://doi.org/10.1002/stem.2345
Coronado D, Godet M, Bourillot P-Y et al (2013) A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Res 10:118–131. https://doi.org/10.1016/j.scr.2012.10.004
Coudreuse D, Nurse P (2010) Driving the cell cycle with a minimal CDK control network. Nature 468:1074–1079. https://doi.org/10.1038/nature09543
Dalton S (2015) Linking the cell cycle to cell fate decisions. Trends Cell Biol 25:592–600. https://doi.org/10.1016/j.tcb.2015.07.007
Desprat R, Thierry-Mieg D, Lailler N et al (2009) Predictable dynamic program of timing of DNA replication in human cells. Genome Res 19:2288–2299. https://doi.org/10.1101/gr.094060.109
Donaldson AD, Raghuraman MK, Friedman KL et al (1998) CLB5-dependent activation of late replication origins in S. cerevisiae. Mol Cell 2:173–182
Donley N, Thayer MJ (2013) DNA replication timing, genome stability and cancer: late and/or delayed DNA replication timing is associated with increased genomic instability. Semin Cancer Biol 23:80–89. https://doi.org/10.1016/j.semcancer.2013.01.001
Ekholm-Reed S, Méndez J, Tedesco D et al (2004) Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol 165:789–800. https://doi.org/10.1083/jcb.200404092
Farrell JA, O’Farrell PH (2014) From egg to gastrula: how the cell cycle is remodeled during the Drosophila mid-blastula transition. Annu Rev Genet 48:269–294. https://doi.org/10.1146/annurev-genet-111212-133531
Fisher DL, Nurse P (1996) A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S-phase and mitosis in the absence of G1 cyclins. EMBO J 15:850–860
Foe VE, Alberts BM (1983) Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J Cell Sci 61:31–70
Fu H, Baris A, Aladjem MI (2018) Replication timing and nuclear structure. Curr Opin Cell Biol 52:43–50. https://doi.org/10.1016/j.ceb.2018.01.004
Harashima H, Dissmeyer N, Schnittger A (2013) Cell cycle control across the eukaryotic kingdom. Trends Cell Biol 23:345–356. https://doi.org/10.1016/j.tcb.2013.03.002
Heichinger C, Penkett CJ, Bähler J, Nurse P (2006) Genome-wide characterization of fission yeast DNA replication origins. EMBO J 25:5171–5179. https://doi.org/10.1038/sj.emboj.7601390
Hendler A, Medina EM, Buchler NE et al (2018) The evolution of a G1/S transcriptional network in yeasts. Curr Genet 64:81–86. https://doi.org/10.1007/s00294-017-0726-3
Hereford LM, Osley MA, Ludwig TR, McLaughlin CS (1981) Cell-cycle regulation of yeast histone mRNA. Cell 24:367–375
Hiratani I, Ryba T, Itoh M et al (2008) Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol 6:e245. https://doi.org/10.1371/journal.pbio.0060245
Hiratani I, Ryba T, Itoh M et al (2010) Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res 20:155–169. https://doi.org/10.1101/gr.099796.109
Homem CCF, Repic M, Knoblich JA (2015) Proliferation control in neural stem and progenitor cells. Nat Rev Neurosci 16:647–659. https://doi.org/10.1038/nrn4021
Hu F, Aparicio OM (2005) Swe1 regulation and transcriptional control restrict the activity of mitotic cyclins toward replication proteins in Saccharomyces cerevisiae. Proc Natl Acad Sci USA 102:8910–8915. https://doi.org/10.1073/pnas.0406987102
Hyrien O, Maric C, Méchali M (1995) Transition in specification of embryonic metazoan DNA replication origins. Science 270:994–997
Kozar K, Ciemerych MA, Rebel VI et al (2004) Mouse development and cell proliferation in the absence of d-cyclins. Cell 118:477–491. https://doi.org/10.1016/j.cell.2004.07.025
Krasinska L, Besnard E, Cot E et al (2008) Cdk1 and Cdk2 activity levels determine the efficiency of replication origin firing in Xenopus. EMBO J 27:758–769. https://doi.org/10.1038/emboj.2008.16
Lange C, Calegari F (2010) Cdks and cyclins link G1 length and differentiation of embryonic, neural and hematopoietic stem cells. Cell Cycle 9:1893–1900. https://doi.org/10.4161/cc.9.10.11598
Lange C, Huttner WB, Calegari F (2009) Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5:320–331. https://doi.org/10.1016/j.stem.2009.05.026
Laskey RA (1985) Chromosome replication in early development of Xenopus laevis. J Embryol Exp Morphol 89 Suppl:285–296
Lengronne A, Schwob E (2002) The yeast CDK inhibitor Sic1 prevents genomic instability by promoting replication origin licensing in late G(1). Mol Cell 9:1067–1078
Macheret M, Halazonetis TD (2018) Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555:112–116. https://doi.org/10.1038/nature25507
Masai H, Yang C-C, Matsumoto S (2017) Mrc1/Claspin: a new role for regulation of origin firing. Curr Genet 63:813–818. https://doi.org/10.1007/s00294-017-0690-y
Morgan DO (1997) Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 13:261–291. https://doi.org/10.1146/annurev.cellbio.13.1.261
Morgan DO (2007) The cell cycle: principles of control. New Science Press, London
Muller CA, Nieduszynski CA (2012) Conservation of replication timing reveals global and local regulation of replication origin activity. Genome Res 22:1953–1962. https://doi.org/10.1101/gr.139477.112
Müller CA, Nieduszynski CA (2017) DNA replication timing influences gene expression level. J Cell Biol 216:1907–1914. https://doi.org/10.1083/jcb.201701061
Murakami H, Keeney S (2014) Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158:861–873. https://doi.org/10.1016/j.cell.2014.06.028
Perrot A, Millington CL, Gómez-Escoda B et al (2018) CDK activity provides temporal and quantitative cues for organizing genome duplication. PLoS Genet 14:e1007214. https://doi.org/10.1371/journal.pgen.1007214
Pope BD, Hiratani I, Gilbert DM (2010) Domain-wide regulation of DNA replication timing during mammalian development. Chromosome Res 18:127–136. https://doi.org/10.1007/s10577-009-9100-8
Pourkarimi E, Bellush JM, Whitehouse I (2016) Spatiotemporal coupling and decoupling of gene transcription with DNA replication origins during embryogenesis in C. elegans. elife. https://doi.org/10.7554/eLife.21728
Rahi SJ, Pecani K, Ondracka A et al (2016) The CDK-APC/C oscillator predominantly entrains periodic cell-cycle transcription. Cell 165:475–487. https://doi.org/10.1016/j.cell.2016.02.060
Rivera-Mulia JC, Buckley Q, Sasaki T et al (2015) Dynamic changes in replication timing and gene expression during lineage specification of human pluripotent stem cells. Genome Res 25:1091–1103. https://doi.org/10.1101/gr.187989.114
Rivera-Mulia JC, Dimond A, Vera D et al (2018) Allele-specific control of replication timing and genome organization during development. Genome Res. https://doi.org/10.1101/gr.232561.117
Rodríguez-Martínez M, Pinzón N, Ghommidh C et al (2017) The gastrula transition reorganizes replication-origin selection in Caenorhabditis elegans. Nat Struct Mol Biol 24:290–299. https://doi.org/10.1038/nsmb.3363
Rustici G, Mata J, Kivinen K et al (2004) Periodic gene expression program of the fission yeast cell cycle. Nat Genet 36:809–817. https://doi.org/10.1038/ng1377
Ryba T, Hiratani I, Lu J et al (2010) Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res 20:761–770. https://doi.org/10.1101/gr.099655.109
Santamaría D, Barrière C, Cerqueira A et al (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448:811–815. https://doi.org/10.1038/nature06046
Siddiqui K, Siddiqui K, On KF et al (2013) Regulating DNA replication in eukarya. Cold Spring Harb Perspect Biol. https://doi.org/10.1101/cshperspect.a012930
Spruck CH, Won KA, Reed SI (1999) Deregulated cyclin E induces chromosome instability. Nature 401:297–300. https://doi.org/10.1038/45836
Swaffer MP, Jones AW, Flynn HR et al (2016) CDK substrate phosphorylation and ordering the cell cycle. Cell 167:1750–1761.e16. https://doi.org/10.1016/j.cell.2016.11.034
Tanaka S, Diffley JFX (2002) Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes Dev 16:2639–2649. https://doi.org/10.1101/gad.1011002
Teixeira LK, Wang X, Li Y et al (2015) Cyclin E deregulation promotes loss of specific genomic regions. Curr Biol 25:1327–1333. https://doi.org/10.1016/j.cub.2015.03.022
Touati SA, Kataria M, Jones AW et al (2018) Phosphoproteome dynamics during mitotic exit in budding yeast. EMBO J. https://doi.org/10.15252/embj.201798745
Walter J, Newport JW (1997) Regulation of replicon size in Xenopus egg extracts. Science 275:993–995
Wilson KA, Elefanty AG, Stanley EG, Gilbert DM (2016) Spatio-temporal re-organization of replication foci accompanies replication domain consolidation during human pluripotent stem cell lineage specification. Cell Cycle 15:2464–2475. https://doi.org/10.1080/15384101.2016.1203492
Wu P-YJ, Nurse P (2009) Establishing the program of origin firing during S phase in fission yeast. Cell 136:852–864. https://doi.org/10.1016/j.cell.2009.01.017
Wu P-YJ, Nurse P (2014) Replication origin selection regulates the distribution of meiotic recombination. Mol Cell 53:655–662. https://doi.org/10.1016/j.molcel.2014.01.022
Yaffe E, Farkash-Amar S, Polten A et al (2010) Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet 6:e1001011. https://doi.org/10.1371/journal.pgen.1001011
Acknowledgements
We thank Damien Coudreuse for critical reading of the manuscript. This work was supported by funding from the Institut National du Cancer (INCA, PLBIO 15-043) and the Région Bretagne. We apologize to any authors whose work was not cited due to space restrictions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Singh, B., Wu, PY.J. Regulation of the program of DNA replication by CDK: new findings and perspectives. Curr Genet 65, 79–85 (2019). https://doi.org/10.1007/s00294-018-0860-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-018-0860-6