
Abstract
Full genome recoding, or rewriting codon meaning, through chemical synthesis of entire bacterial chromosomes has become feasible in the past several years. Recoding an organism can impart new properties including non-natural amino acid incorporation, virus resistance, and biocontainment. The estimated cost of construction that includes DNA synthesis, assembly by recombination, and troubleshooting, is now comparable to costs of early stage development of drugs or other high-tech products. Here, we discuss several recently published assembly methods and provide some thoughts on the future, including how synthetic efforts might benefit from the analysis of natural recoding processes and organisms that use alternative genetic codes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: Why recode organisms?
Human manipulation of the genetic code began in the 1960s as molecular geneticists isolated nonsense and missense suppressor mutations. There, tRNAs were altered to insert “incorrect” amino acids at certain positions in proteins, but such mutations lead to an ambiguous code generating variable products and inefficient protein production (Kaplan 1971; Rogers et al. 1992). Recently, genome-scale modification of the genetic code has become feasible, which could enable construction of organisms with unambiguous alternative genetic codes.
A specific codon can be replaced with a synonymous one in the degenerate 64-codon genetic code (Plotkin and Kudla 2011). Done globally with corresponding tRNA removal, this entirely removes a codon from the genome, allowing reassignment for another use (which may be no use). Recoding, or changing a codon’s use in a genome, has been observed naturally in dozens of organisms, but often for stop codons (Ivanova et al. 2014; Ling et al. 2015). By synthetically recoding organisms, we can gain several valuable features (Lajoie et al. 2016; Mukai et al. 2017).
Repurpose codons for non-natural amino acids
With a free codon and tRNA available, non-natural amino acids could be introduced at an unprecedented ~100% incorporation efficiency. Already, tRNA engineering has enabled incorporation of non-natural amino acids into proteins (Dumas et al. 2015; Wang et al. 2014; Young and Schultz 2010), but efficiency is limited due to competing natural translation processes. New amino acids may improve and even expand protein functions (Wang et al. 2006; Xiao et al. 2015), such as by fluorination (Marsh 2014). A novel proteomic signature would also help in identifying escaped engineered organisms.
Virus resistance
In industrial fermentation, virus contamination is a significant issue: entire production runs can be lost because of a bacteriophage (Jones et al. 2000) and is a longstanding concern for dairy industry lactic acid bacteria (Garneau and Moineau 2011; Samson and Moineau 2013). Recoded cells with specific tRNAs removed or used for a novel amino acid should be broadly resistant to decoding infective nucleic acid messages, such as from viruses. A bacterial strain that cannot recognize a common sense codon should be unable to translate essentially any phage gene.
Resistance to horizontal gene transfer
A general problem for the release of engineered microbes into the wild is that, unlike higher animals and plants, microbes readily exchange DNA with each other across species barriers. Synthetic biologists have envisioned and constructed bacteria to decontaminate pesticide-contaminated fields (Mattozzi and Keasling 2010), non-invasively diagnose the presence of chemicals in the gut (Kotula et al. 2014; Riglar et al. 2017), or photosynthetically synthesize biofuels in open ponds (Savage et al. 2008). Such organisms could exchange DNA with other unengineered microbes, with unpredictable environmental consequences. Recoding can block functional horizontal gene transfer: reassigning stop codons as sense and inserting throughout coding sequences would make recoded host genes unreadable by most other microbes, and removing sense codons would make foreign DNA unreadable in the recoded host.
Biocontainment
Repurposing codons for non-natural amino acids also allows for the development of improved auxotrophs. Synthetic amino acids not found in Nature can be inserted into some essential genes, ensuring inability to survive without the non-natural amino acid feedstock. This could create a realistic version of Michael Crichton’s “lysine contingency” biocontainment in Jurassic Park (New York: Ballantine Books, 1990). Another potential strategy uses a toxin to prevent DNA transfer from engineered organism to environmental neighbors. Adding a non-recoded broad-range toxin sequence (ex., endonuclease) to a transgenic cassette, the recoded host cannot express the lethal gene while other organisms acquiring the cassette can. The toxin also selects against reacquiring native tRNA machinery repurposed when recoding.
New/improved functions and genome reduction
Since recoding methods involve entire genome synthesis (discussed below), new gene clusters can be concurrently inserted, along with deletions for genome reduction. Many genes needed to adapt in uncertain, changing environments (Hutchison et al. 2016) are unneeded in controlled settings such as bioreactors. For one-trick pony industrial strains, the compacted genome itself could improve stability (Csorgo et al. 2012).
Learn fundamental biology
Besides engineering applications, recoding can be a platform to address biological questions. Studying cell response to massive codon replacements, new properties related to global transcription/translation mechanisms may emerge. Recoding of viruses has led to the elucidation of mechanisms by comparison with native virus to identify key sequences and codon usage properties (Martinez et al. 2016). Additionally, non-natural amino acid labeling can selectively tag all proteins of a pathway, enabling systems-level mechanistic studies.
Assembly of recoded organisms: recent efforts
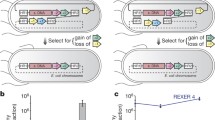
Only several genome-wide recoding efforts have been published. While major advances have been presented for viruses (Coleman et al. 2008, 2011; Martinez et al. 2016) and yeast (discussed briefly), this section focuses on efforts recoding bacteria (Escherichia coli and Salmonella). Assembly methods can be classified into three categories: (1) editing the existing genome, (2) rebuilding by segments, and (3) complete de novo construction (Fig. 1).

Recoding assembly strategies. Current recoding methods can be categorized as a editing the existing genome, b rebuilding by segments, and c complete de novo synthesis. a Site-specific point mutations are made throughout the native genome to change target codons, using oligonucleotides. b The native genome is rebuilt in the native host organism through an iterative stepwise procedure with synthetic DNA segments containing the recoding changes. Shown, segments are integrated by homologous recombination. As in (a), different sections can be built separately and combined in a single strain downstream. c An entire genome is made de novo from synthesized fragments and assembled in one pot, bypassing the need for the native genome (and maybe organism). Larger fragments are successively built up from smaller ones
Editing existing: E. coli TAG recoding by MAGE and CAGE (2011–2013)
A first success was George Church’s lab using multiplex automated genome engineering (MAGE) to change all 321 TAG stop codons to TAA in E. coli (Isaacs et al. 2011). In this method, short oligonucleotides are used to make site-specific codon changes through recombination events (Fig. 1a). The strategy made 10 changes per strain in parallel across 32 strains and combined the results using bacterial conjugation, termed conjugative assembly genome engineering (CAGE). Completion of the TAG to TAA recoded E. coli (“rE.coli”) strain included deletion of release factor RF1, which recognizes the UAG stop (Lajoie et al. 2013b).
rE.coli was shown to indeed have recoded advantages of (1) virus resistance (Ma and Isaacs 2016) and (2) was further engineered for non-natural phenylalanine-derivative amino acid incorporation to (3) create auxotrophic strains dependent on supplied synthetic amino acid, with undetectable escape rates of less than 1 in 1012 for effective biocontainment (Rovner et al. 2015).
MAGE-based approaches were also used to look at viability consequences of recoding essential genes (Lajoie et al. 2013a; Napolitano et al. 2016), because of the importance of codon usage bias in controlling aspects of gene regulation (Goodman et al. 2013; Quax et al. 2015; Tuller et al. 2010a, b).
Rebuilding by segments: integrase-based 50 kb fragments in E. coli (2016)
Moving from MAGE, the Church Lab developed another method involving complete synthesis and lambda phage integrase recombination (Ostrov et al. 2016). They also created and used design software to automate the recoded genome blueprint. Entire 50-kb segments of recoded DNA were synthesized de novo in 2–4 kb fragments and combined in yeast with a plasmid backbone. This backbone has an attP integrase site for integration into a strain modified with a corresponding target attB site in a multi-step process. This method was used to reduce from 64 to 57 codons (over 62,000 replacements for “rE.coli-57”) across 87 strains, with the problem of final hierarchical assembly still a work-in-progress.
Rebuilding by segments: testing replacement schemes, REXER in E. coli (2016)
An assembly method from Jason Chin and colleagues—named replicon excision for enhanced genome engineering through reprogrammed recombination (REXER)—uses the larger bacterial artificial chromosome (BAC) for 100-kb segment replacements in an iterative stepping process (Fig. 1b), also using yeast-assisted assembly of synthetic recoded DNA fragments (Wang et al. 2016). The recoded section is excised by Cas9 after transformation and integrated into the genome by lambda Red homologous recombination. In addition, Wang et al. demonstrate a troubleshooting technique for unviable or poorly growing strains due to recoding. Their efforts highlight the major difficulty that many designed synonymous replacement schemes will be unviable, even on a small scale.
Rebuilding by segments: Salmonella leucine recoding using SIRCAS (2017)
A method we (Pamela Silver Lab) developed also uses homologous recombination and tiled antibiotic resistance marker stepping, shown to make 1557 synonymous leucine replacements across 176 genes in Salmonella typhimurium (Lau et al. 2017). Named SIRCAS for stepwise integration of rolling circle amplification segments, the method uses 10–25 kb linear fragments of synthetic DNA obtained from rolling circle amplification of constructs assembled in yeast. This method requires only an initial genomic integration of inducible lambda Red recombination genes, allowing a rapid two-day turnaround for recoded segment integration.
Complete de novo synthesis: a minimal genome in Mycoplasma (2016)
Though not a codon reassignment effort, the major achievement of creating a minimal genome for the already efficient Mycoplasma mycoides (Hutchison et al. 2016) presents an alternative assembly method. Using massive construction from oligonucleotides to assemble increasingly larger fragments (Fig. 1c), the genome was reduced from 1079 to 531 kb. The herculean procedure used an expansion and contraction pragmatic approach, knowledge of essential genes and a Tn5 transposon disruption map. Amazingly, the newly synthesized genomes were introduced into Mycoplasma that then replicated the genome to yield viable strains.
Rebuilding by segments: synthetic yeast chromosomes (2017)
The Synthetic Yeast Genome Project is a huge effort across many organizations to completely build yeast chromosomes from scratch. A set of seven papers published in Science (March 10, 2017) describe construction of five complete chromosomes, which included recoding TAG to TAA stop codons and deleting all tRNAs, to be moved to a tRNA-only chromosome (Richardson et al. 2017). Their methods use yeast’s natural homologous recombination to integrate 30–60 kb segments of recoded DNA, similar to iterative segmented-rebuilds of bacteria. Notably, the yeast project also includes a troubleshooting strategy (Mitchell et al. 2017) that may be useful for bacterial efforts. Recoding methods for yeast can augment yeast genetics studies useful for industrial purposes (Cubillos 2016; Snoek et al. 2016).
Comparison of methods
Likely the best recoding approach will incorporate aspects from several methods. All have a similar global strategy of evaluating partially recoded strains for viability before piecewise assembly into a single organism. Notably, methods are interchangeable in that recoded DNA can be taken from a viable strain and transferred to another, such as using REXER to combine 50 kb sections of rE.coli-57 precursors or 100 kb sections recoded by SIRCAS. The MAGE-based methods to make a handful of changes in a single strain may be useful in later stages of recoding or in adjusting unviable designs (Ostrov et al. 2016). Strain parallelization in each method gives the possibility of rapid construction.
Construction methods are in place, but troubleshooting methods all require a laborious process filled with trial-and-error. Though groups have tried identifying canonical “rules” for sense codon recoding, many of the empirically found guidelines might only apply to those specific sequences/organisms. A robust troubleshooting process would be a major lift to the field and is an essential part of the assembly process. In addition, improved speed (and cost) in high fidelity DNA synthesis would be a huge boost toward fully recoding organisms at the megabase scale.
The future
While several powerful assembly methods have been described, we have only had a glimpse of the properties so attractive in theory. Strains with greater instances of codon replacements are needed to truly attain these properties. For example, many infective messages may not contain the TAG stop targeted in recoding, or viruses may adapt (Ivanova et al. 2014). Promisingly, many partially recoded strains discussed have similar overall growth as wild-type versions or could have reduced fitness improved through evolution (Wannier et al. 2017).
Lessons from natural recoding?
A deeper understanding of evolutionary events in natural genome recoding may reveal new evolution-based strategies to complement the rebuild recoding methods developed to date. Recoding has been observed over twenty times throughout the tree of life (Knight et al. 2001; Ling et al. 2015). Many of these organisms are bacteria with reduced genome sizes and/or AT-rich composition, with theories that events leading to these properties resulted in recoding (McCutcheon et al. 2009; Osawa et al. 1992). Similar mechanisms have been proposed for mitochondria, where across species eight sense and all three stop codons are reassigned (Sengupta et al. 2007), often several together in the compact genomes (Adams and Palmer 2003). While this may suggest a role of using genome reduction in synthetic recoding, these evolutionary mechanisms based on altered global genome properties are likely not effective on the rapid time scales desired.
However, codon reassignments in large eukaryotic genomes—as in yeasts [4–8 K genes, 9–19 Mb (Riley et al. 2016)]—likely required codon-specific selective pressure. In two separate yeast clades, leucine codon CTG is reassigned to translate as either alanine or serine. Species diverging prior to the predicted recoding event contain thousands of CTG positions in coding regions (Riley et al. 2016) that are not conserved in recoded species (Muhlhausen and Kollmar 2014). These CTGs were proposed to be systematically disfavored and driven to rarity by “mischarging” of tRNACAG, which happens in extant yeasts such as Candida albicans (Massey et al. 2003), or an inability to translate CTG efficiently due to the loss of tRNACAG (Muhlhausen et al. 2016). A more thorough analysis and identification of recoded species lineages may uncover evolutionary paths that inspire synthetic efforts.
Even without new evolutionary insights, a pragmatic option may be to apply selection pressures against a specific codon in an experimental setup, to mirror natural evolution toward reassignment. Usage of a specific codon might be disfavored by introducing a competing tRNA isotype to increase missense errors (Ruan et al. 2008; Santos et al. 1999) or impairing translation by deleting tRNA genes (Bloom-Ackermann et al. 2014). Such pressure may allow recoding instances through non-synonymous routes while maintaining viability, potentially fixing unviable strains in whole-rebuild methods.
Other natural examples may inspire more original strategies. In select ciliates, all three stop codons have added sense meanings (Heaphy et al. 2016; Swart et al. 2016), another possible expansion strategy. Instead of being permanently encoded genome-wide, it may be worth recoding in real-time: Mycobacterium bovis uses a hypoxic stress-induced tRNA modification coupled with a distinctly codon-biased set of stress response genes to enter a state of dormancy (Chionh et al. 2016). Bacteria Acetohalobium arabaticum dynamically expands its code to incorporate pyrrolysine when grown with trimethylamine (Prat et al. 2012). Perhaps an inducible system can be designed where only some genes are recoded under certain conditions.
Economics of genome recoding and DNA synthesis
We estimate the total cost of recoding an E. coli-sized bacterial genome (~5 Mb) to be a few million US dollars. This includes raw DNA synthesis plus assembly into large pieces and incorporation by stepwise replacement. The price tag should be considered with the potential benefits in a multi-billion-dollar fermentation industry (Erickson and Winters 2012). Growing genome-scale recoding efforts could fundamentally change the economics of DNA synthesis. Large-scale orders for recoded genomes are easy to conceptualize, can be designed computationally based on annotated genome sequences, and can be ordered at scale, minimizing processing costs. In contrast, conceptualization and rational design of a multi-component genetic circuit of even 10 kb can still be intellectually prohibitive. We see completion of genome recoding efforts as playing a key role in driving down DNA synthesis costs by increasing demand.
Final remarks
Along with recoding current industrial strains, the promise of synthetic genome recoding is to create versatile, genetically isolated base strains on which to build desired functions. Methods are now in place to fully recode a sense codon in bacteria, with major hurdles being a more robust troubleshooting method for non-viable designs and the still unknown effects of such large-scale codon replacements. Since millions of dollars may be prohibitively expensive for one lab, academic recoding may benefit from large-scale collective efforts, such as in the Yeast Synthetic Genome Project, as the process is so amenable to partitioning.
References
Adams KL, Palmer JD (2003) Evolution of mitochondrial gene content: gene loss and transfer to the nucleus. Mol Phylogenet Evol 29:380–395
Bloom-Ackermann Z, Navon S, Gingold H, Towers R, Pilpel Y, Dahan O (2014) A comprehensive tRNA deletion library unravels the genetic architecture of the tRNA pool. PLoS Genet 10:e1004084. doi:10.1371/journal.pgen.1004084
Chionh YH, McBee M, Babu IR, Hia F, Lin W, Zhao W, Cao J, Dziergowska A, Malkiewicz A, Begley TJ, Alonso S, Dedon PC (2016) tRNA-mediated codon-biased translation in mycobacterial hypoxic persistence. Nat Commun 7:13302. doi:10.1038/ncomms13302
Coleman JR, Papamichail D, Skiena S, Futcher B, Wimmer E, Mueller S (2008) Virus attenuation by genome-scale changes in codon pair bias. Science 320:1784–1787. doi:10.1126/science.1155761
Coleman JR, Papamichail D, Yano M, Garcia-Suarez Mdel M, Pirofski LA (2011) Designed reduction of Streptococcus pneumoniae pathogenicity via synthetic changes in virulence factor codon-pair bias. J Infect Dis 203:1264–1273. doi:10.1093/infdis/jir010
Csorgo B, Feher T, Timar E, Blattner FR, Posfai G (2012) Low-mutation-rate, reduced-genome Escherichia coli: an improved host for faithful maintenance of engineered genetic constructs. Microb Cell Fact 11:11. doi:10.1186/1475-2859-11-11
Cubillos FA (2016) Exploiting budding yeast natural variation for industrial processes. Curr Genet 62:745–751. doi:10.1007/s00294-016-0602-6
Dumas A, Lercher L, Spicer CD, Davis BG (2015) Designing logical codon reassignment—expanding the chemistry in biology. Chem Sci 6:50–69. doi:10.1039/c4sc01534g
Erickson B, Winters P (2012) Perspective on opportunities in industrial biotechnology in renewable chemicals. Biotechnol J 7:176–185. doi:10.1002/biot.201100069
Garneau JE, Moineau S (2011) Bacteriophages of lactic acid bacteria and their impact on milk fermentations. Microb Cell Fact 10(Suppl 1):S20. doi:10.1186/1475-2859-10-S1-S20
Goodman DB, Church GM, Kosuri S (2013) Causes and effects of N-terminal codon bias in bacterial genes. Science 342:475–479. doi:10.1126/science.1241934
Heaphy SM, Mariotti M, Gladyshev VN, Atkins JF, Baranov PV (2016) Novel ciliate genetic code variants including the reassignment of all three stop codons to sense codons in condylostoma magnum. Mol Biol Evol 33:2885–2889. doi:10.1093/molbev/msw166
Hutchison CA 3rd, Chuang RY, Noskov VN, Assad-Garcia N, Deerinck TJ, Ellisman MH, Gill J, Kannan K, Karas BJ, Ma L, Pelletier JF, Qi ZQ, Richter RA, Strychalski EA, Sun L, Suzuki Y, Tsvetanova B, Wise KS, Smith HO, Glass JI, Merryman C, Gibson DG, Venter JC (2016) Design and synthesis of a minimal bacterial genome. Science 351:aad6253. doi:10.1126/science.aad6253
Isaacs FJ, Carr PA, Wang HH, Lajoie MJ, Sterling B, Kraal L, Tolonen AC, Gianoulis TA, Goodman DB, Reppas NB, Emig CJ, Bang D, Hwang SJ, Jewett MC, Jacobson JM, Church GM (2011) Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333:348–353. doi:10.1126/science.1205822
Ivanova NN, Schwientek P, Tripp HJ, Rinke C, Pati A, Huntemann M, Visel A, Woyke T, Kyrpides NC, Rubin EM (2014) Stop codon reassignments in the wild. Science 344:909–913. doi:10.1126/science.1250691
Jones DT, Shirley M, Wu X, Keis S (2000) Bacteriophage infections in the industrial acetone butanol (AB) fermentation process. J Mol Microbiol Biotechnol 2:21–26
Kaplan S (1971) Lysine suppressor in Escherichia coli. J Bacteriol 105:984–987
Knight RD, Freeland SJ, Landweber LF (2001) Rewiring the keyboard: evolvability of the genetic code. Nat Rev Genet 2:49–58. doi:10.1038/35047500
Kotula JW, Kerns SJ, Shaket LA, Siraj L, Collins JJ, Way JC, Silver PA (2014) Programmable bacteria detect and record an environmental signal in the mammalian gut. Proc Natl Acad Sci USA 111:4838–4843. doi:10.1073/pnas.1321321111
Lajoie MJ, Kosuri S, Mosberg JA, Gregg CJ, Zhang D, Church GM (2013a) Probing the limits of genetic recoding in essential genes. Science 342:361–363. doi:10.1126/science.1241460
Lajoie MJ, Rovner AJ, Goodman DB, Aerni HR, Haimovich AD, Kuznetsov G, Mercer JA, Wang HH, Carr PA, Mosberg JA, Rohland N, Schultz PG, Jacobson JM, Rinehart J, Church GM, Isaacs FJ (2013b) Genomically recoded organisms expand biological functions. Science 342:357–360. doi:10.1126/science.1241459
Lajoie MJ, Soll D, Church GM (2016) Overcoming challenges in engineering the genetic code. J Mol Biol 428:1004–1021. doi:10.1016/j.jmb.2015.09.003
Lau YH, Stirling F, Kuo J, Karrenbelt MAP, Chan YA, Riesselman A, Horton CA, Schafer E, Lips D, Weinstock MT, Gibson DG, Way JC, Silver PA (2017) Large-scale recoding of a bacterial genome by iterative recombineering of synthetic DNA. Nucleic Acids Res. doi:10.1093/nar/gkx415
Ling J, O’Donoghue P, Soll D (2015) Genetic code flexibility in microorganisms: novel mechanisms and impact on physiology. Nat Rev Microbiol 13:707–721. doi:10.1038/nrmicro3568
Ma NJ, Isaacs FJ (2016) Genomic recoding broadly obstructs the propagation of horizontally transferred genetic elements. Cell Syst 3:199–207. doi:10.1016/j.cels.2016.06.009
Marsh EN (2014) Fluorinated proteins: from design and synthesis to structure and stability. Acc Chem Res 47:2878–2886. doi:10.1021/ar500125m
Martinez MA, Jordan-Paiz A, Franco S, Nevot M (2016) Synonymous virus genome recoding as a tool to impact viral fitness. Trends Microbiol 24:134–147. doi:10.1016/j.tim.2015.11.002
Massey SE, Moura G, Beltrao P, Almeida R, Garey JR, Tuite MF, Santos MA (2003) Comparative evolutionary genomics unveils the molecular mechanism of reassignment of the CTG codon in Candida spp. Genome Res 13:544–557. doi:10.1101/gr.811003
Mattozzi MDLP, Keasling JD (2010) Rationally engineered biotransformation of p-nitrophenol. Biotechnol Prog 26:616–621. doi:10.1002/btpr.382
McCutcheon JP, McDonald BR, Moran NA (2009) Origin of an alternative genetic code in the extremely small and GC-rich genome of a bacterial symbiont. PLoS Genet 5:e1000565. doi:10.1371/journal.pgen.1000565
Mitchell LA, Wang A, Stracquadanio G, Kuang Z, Wang X, Yang K, Richardson S, Martin JA, Zhao Y, Walker R, Luo Y, Dai H, Dong K, Tang Z, Yang Y, Cai Y, Heguy A, Ueberheide B, Fenyo D, Dai J, Bader JS, Boeke JD (2017) Synthesis, debugging, and effects of synthetic chromosome consolidation: synVI and beyond. Science. doi:10.1126/science.aaf4831
Muhlhausen S, Kollmar M (2014) Molecular phylogeny of sequenced Saccharomycetes reveals polyphyly of the alternative yeast codon usage. Genome Biol Evol 6:3222–3237
Muhlhausen S, Findeisen P, Plessmann U, Urlaub H, Kollmar M (2016) A novel nuclear genetic code alteration in yeasts and the evolution of codon reassignment in eukaryotes. Genome Res 26:945–955. doi:10.1101/gr.200931.115
Mukai T, Lajoie MJ, Englert M, Soll D (2017) Rewriting the genetic code. Annu Rev Microbiol. doi:10.1146/annurev-micro-090816-093247
Napolitano MG, Landon M, Gregg CJ, Lajoie MJ, Govindarajan L, Mosberg JA, Kuznetsov G, Goodman DB, Vargas-Rodriguez O, Isaacs FJ, Soll D, Church GM (2016) Emergent rules for codon choice elucidated by editing rare arginine codons in Escherichia coli. Proc Natl Acad Sci USA 113:E5588–E5597. doi:10.1073/pnas.1605856113
Osawa S, Jukes TH, Watanabe K, Muto A (1992) Recent evidence for evolution of the genetic code. Microbiol Rev 56:229–264
Ostrov N, Landon M, Guell M, Kuznetsov G, Teramoto J, Cervantes N, Zhou M, Singh K, Napolitano MG, Moosburner M, Shrock E, Pruitt BW, Conway N, Goodman DB, Gardner CL, Tyree G, Gonzales A, Wanner BL, Norville JE, Lajoie MJ, Church GM (2016) Design, synthesis, and testing toward a 57-codon genome. Science 353:819–822. doi:10.1126/science.aaf3639
Plotkin JB, Kudla G (2011) Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 12:32–42. doi:10.1038/nrg2899
Prat L, Heinemann IU, Aerni HR, Rinehart J, O’Donoghue P, Soll D (2012) Carbon source-dependent expansion of the genetic code in bacteria. Proc Natl Acad Sci USA 109:21070–21075. doi:10.1073/pnas.1218613110
Quax TE, Claassens NJ, Soll D, van der Oost J (2015) Codon bias as a means to fine-tune gene expression. Mol Cell 59:149–161. doi:10.1016/j.molcel.2015.05.035
Richardson SM, Mitchell LA, Stracquadanio G, Yang K, Dymond JS, DiCarlo JE, Lee D, Huang CL, Chandrasegaran S, Cai Y, Boeke JD, Bader JS (2017) Design of a synthetic yeast genome. Science 355:1040–1044. doi:10.1126/science.aaf4557
Riglar DT, Giessen TW, Baym M, Kerns SJ, Niederhuber MJ, Bronson RT, Kotula JW, Gerber GK, Way JC, Silver PA (2017) Engineered bacteria can function in the mammalian gut long-term as live diagnostics of inflammation. Nat Biotechnol 35:653–658. doi:10.1038/nbt.3879
Riley R, Haridas S, Wolfe KH, Lopes MR, Hittinger CT, Goker M, Salamov AA, Wisecaver JH, Long TM, Calvey CH, Aerts AL, Barry KW, Choi C, Clum A, Coughlan AY, Deshpande S, Douglass AP, Hanson SJ, Klenk HP, LaButti KM, Lapidus A, Lindquist EA, Lipzen AM, Meier-Kolthoff JP, Ohm RA, Otillar RP, Pangilinan JL, Peng Y, Rokas A, Rosa CA, Scheuner C, Sibirny AA, Slot JC, Stielow JB, Sun H, Kurtzman CP, Blackwell M, Grigoriev IV, Jeffries TW (2016) Comparative genomics of biotechnologically important yeasts. Proc Natl Acad Sci USA 113:9882–9887. doi:10.1073/pnas.1603941113
Rogers MJ, Adachi T, Inokuchi H, Soll D (1992) Switching tRNA(Gln) identity from glutamine to tryptophan. Proc Natl Acad Sci USA 89:3463–3467
Rovner AJ, Haimovich AD, Katz SR, Li Z, Grome MW, Gassaway BM, Amiram M, Patel JR, Gallagher RR, Rinehart J, Isaacs FJ (2015) Recoded organisms engineered to depend on synthetic amino acids. Nature 518:89–93. doi:10.1038/nature14095
Ruan B, Palioura S, Sabina J, Marvin-Guy L, Kochhar S, Larossa RA, Soll D (2008) Quality control despite mistranslation caused by an ambiguous genetic code. Proc Natl Acad Sci USA 105:16502–16507. doi:10.1073/pnas.0809179105
Samson JE, Moineau S (2013) Bacteriophages in food fermentations: new frontiers in a continuous arms race. Annu Rev Food Sci Technol 4:347–368. doi:10.1146/annurev-food-030212-182541
Santos MA, Cheesman C, Costa V, Moradas-Ferreira P, Tuite MF (1999) Selective advantages created by codon ambiguity allowed for the evolution of an alternative genetic code in Candida spp. Mol Microbiol 31:937–947
Savage DF, Way J, Silver PA (2008) Defossiling fuel: how synthetic biology can transform biofuel production. ACS Chem Biol 3:13–16. doi:10.1021/cb700259j
Sengupta S, Yang X, Higgs PG (2007) The mechanisms of codon reassignments in mitochondrial genetic codes. J Mol Evol 64:662–688. doi:10.1007/s00239-006-0284-7
Snoek T, Verstrepen KJ, Voordeckers K (2016) How do yeast cells become tolerant to high ethanol concentrations? Curr Genet 62:475–480. doi:10.1007/s00294-015-0561-3
Swart EC, Serra V, Petroni G, Nowacki M (2016) Genetic codes with no dedicated stop codon: context-dependent translation termination. Cell 166:691–702. doi:10.1016/j.cell.2016.06.020
Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y (2010a) An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell 141:344–354. doi:10.1016/j.cell.2010.03.031
Tuller T, Waldman YY, Kupiec M, Ruppin E (2010b) Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA 107:3645–3650. doi:10.1073/pnas.0909910107
Wang L, Xie J, Schultz PG (2006) Expanding the genetic code. Annu Rev Biophys Biomol Struct 35:225–249. doi:10.1146/annurev.biophys.35.101105.121507
Wang K, Sachdeva A, Cox DJ, Wilf NM, Lang K, Wallace S, Mehl RA, Chin JW (2014) Optimized orthogonal translation of unnatural amino acids enables spontaneous protein double-labelling and FRET. Nat Chem 6:393–403. doi:10.1038/nchem.1919
Wang K, Fredens J, Brunner SF, Kim SH, Chia T, Chin JW (2016) Defining synonymous codon compression schemes by genome recoding. Nature 539:59–64. doi:10.1038/nature20124
Wannier TM, Kunjapur AM, Rice DP, McDonald MJ, Desai MM, Church GM (2017) Long-term adaptive evolution of genomically recoded Escherichia coli. bioRxiv. doi:10.1101/162834
Xiao H, Nasertorabi F, Choi SH, Han GW, Reed SA, Stevens RC, Schultz PG (2015) Exploring the potential impact of an expanded genetic code on protein function. Proc Natl Acad Sci USA 112:6961–6966. doi:10.1073/pnas.1507741112
Young TS, Schultz PG (2010) Beyond the canonical 20 amino acids: expanding the genetic lexicon. J Biol Chem 285:11039–11044. doi:10.1074/jbc.R109.091306
Acknowledgements
We thank Matthew Weinstock and Daniel Gibson (SGI), and George Church, Nili Ostrov, Matthieu Landon, Cameron Gardner and other Church Lab members (Harvard Medical School), for previous stimulating discussions. We acknowledge support from Defense Advanced Research Projects Agency Grant HR0011-15-C-0094; the National Institute of General Medical Sciences of the NIH for a Ruth L. Kirschstein National Research Service Award (F32GM125108 to J.K.) and an NIH training grant (5T32GM007598, F.S.); and the Wellcome Trust for a Sir Henry Wellcome postdoctoral fellowship (107402/Z/15/Z to Y.H.L.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Kuo, J., Stirling, F., Lau, Y.H. et al. Synthetic genome recoding: new genetic codes for new features. Curr Genet 64, 327–333 (2018). https://doi.org/10.1007/s00294-017-0754-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-017-0754-z