
Abstract
Ustilaginoidea virens is the causal agent of rice false smut disease resulting in quantitative and qualitative losses in rice. To gain insights into the pathogenic mechanisms of U. virens, we established a T-DNA insertion mutant library of U. virens through Agrobacterium tumefaciens-mediated transformation and selected an enhanced pathogenicity mutant (i.e., B3277). We analyzed the biological characteristics of the wild-type P1 and B3277. The growth rate and sporulation of B3277 were decreased compared with those of P1; the ferrous iron could be utilized by B3277, but inhibited the growth of P1. Southern blot analysis was performed to verify the copy number of the foreign gene inserted in the genomic DNA and only one copy of the T-DNA was found. The combined hiTAIL-PCR with RACE-PCR analysis showed the successful cloning of full length of the T-DNA flanking gene associated with pathogenicity, named Uvt3277. Gene expression was analyzed using real-time PCR. Results revealed that Uvt3277 was expressed at lower levels in B3277 than in P1. This gene was then subjected to bioinformatics analysis. The encoded protein of Uvt3277 exhibited high homology with low-affinity iron transporter proteins in some fungi. Transformation of the RNAi vector by constructing the hairpin RNA of the target gene was confirmed as successful. The pathogenicity of the transformant also increased. These results suggested that Uvt3277 may have an important function associated with the pathogenesis of U. virens. This study provides insights into the pathogenic mechanism of U. virens and a molecular target of disease control.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rice false smut (RFS) is caused by the fungus Ustilaginoidea virens. (Che.) Tak. (teleomorph Villosiclava virens) (Tanaka et al. 2008; White et al. 2000). Ustilaginoidea virens infects rice spikelets and converts grains into smut balls whose color changes from yellowish to yellowish orange, green, olive green and ultimately to greenish black (Ashizawa et al. 2012; Tang et al. 2013; Hu et al. 2014). Green false smut balls with numerous chlamydospores contain mycotoxins (ustilaginoidins and ustiloxins), which are poisonous to humans and animals; these toxins inhibit microtubule assembly and promote the cytotoxic-induced formation of antimitotic cytoskeleton (Fan et al. 2012; Yin et al. 2012). False smut disease thus not only reduces grain yield, but also contaminates rice grains and straw. In recent years, RFS has become one of the most devastating rice grain diseases worldwide caused by several factors, such as widespread cultivation of hybrid varieties, heavy application of nitrogenous fertilizers, and climate changes (Zhou et al. 2008), and has attracted more and more attention of researchers.
Studies on rice false smut have mainly focused on the phylogenetic relationship of pathogens, morphological characteristics, diversity of pathogens and ustiloxins, artificial inoculation pathogen transformation, and rice resistance to fungal diseases (Ashizawa et al. 2011; Guo et al. 2012; Sun et al. 2013; Yu et al. 2015; Meng et al. 2015; Andargie and Li 2016). Despite a large number of studies on fungal infections, only a few reports have elucidated the molecular genetics of U. virens, thereby limiting the in-depth understanding of infection and pathogenic mechanisms of this plant pathogen. Moreover, the mechanism of U. virens pathogenesis is largely unknown.
Random insertional mutagenesis and targeted gene disruption have been performed to investigate fungal diseases. For example, Agrobacterium tumefaciens-mediated transformation (ATMT) is conducted to identify genes required for fungal development or infection processes (de Groot et al. 1998; Jiang et al. 2013; Michielse et al. 2005). ATMT in the fungus Verticillium dahliae is effectively used to identify genes associated with pathogenicity and other functions, including VdEg-1 encoding endoglucanase 1 and hydroxyl-methyl glutaryl-CoA synthase VdHMGS (Maruthachalam et al. 2011). Conducting large-scale insertional mutagenesis in Fusarium oxysporum (Michielse et al. 2009; Mullins et al. 2001) contributes further insights into molecular pathogenesis. Magnaporthe oryzae is another excellent model organism for studies involving plant–fungal pathogen interactions (Ebbole 2007; Talbot 2003; Valent 1990) and a series of pathogenicity-related genes have been cloned in M. oryzae. DES1 regulates counter-defenses against host basal resistance (Chi et al. 2009). MoLdb1 involves in the regulation of cell wall proteins, conidiation, sexual development, appressorium formation, and pathogenicity (Li et al. 2010). Molecular technique-based studies are also conducted to detect U. virens (Ashizawa et al. 2010; Zhou et al. 2003). Several genes associated with pathogenicity are isolated through ATMT. A novel promoter, puv880, which may play a central role in the regulation of gene expression is useful for understanding the conidiation and mycelium growth of U. virens (Hu et al. 2013). UvSUN2 might be a virulence factor and possibly required for proper fungal growth, cell wall construction, and stress responses in U. virens (Yu et al. 2015). Moreover, effectors often play essential roles in host–pathogen coevolutionary interactions. Based on genome sequence (Zhang et al. 2014), putative fungal effectors with specific structural motifs in U. virens are found and could induce cell death or defense machinery in nonhost and host plants (Fang et al. 2016).
In our previous studies, we optimized ATMT to establish a T-DNA insertion mutant library of U. virens (Yu et al. 2015) and obtained the enhanced virulence mutant B3277. Here, we cloned the gene inserted by T-DNA. The gene exhibited high homology with low-affinity iron transporter proteins. We also validated that the gene acts in the regulation of hyphal growth and conidiation, and plays an important role in U. virens pathogenicity. This study provides insights into the pathogenic mechanism of U. virens and is a potential molecular target for disease control.
Materials and methods
Fungal strains and growth conditions
Two U. virens strains were used in this study. The wild-type strain was designated as P1 and isolated from field-infected rice. A T-DNA insertion mutant library of U. virens was established through ATMT. Multiple virulence tests were conducted in the field, and the enhanced pathogenic mutant B3277 strain was screened. Two strains were preserved as monoconidial culture in 20 % glycerol at −70 °C. The fungi were grown and maintained in PSA medium (200 g/L potato, 20 g/L sucrose, and 12 g/L agar powder). The material was revitalized in PSA. The rice false smut-susceptible variety, namely, Liangyoupeijiu, was used as experimental rice.
Analysis of the biological traits of the mutant B3277 strain
The mutant B3277 and wild-type P1 strains were cultured in a PSA medium to determine their growth rates. The samples were obtained from the edge of the colony growth using a puncher with a 3.5 mm diameter. These samples were inoculated in the PSA, TB3, and minimum medium (MM) with three replicates for each culture. The colonies were cultured in the dark at 28 °C for 14 days. Then the colonies were photographed and their diameters measured.
The sporulation capacity was analyzed according to the following procedures. Six dishes containing 50 mL of liquid PS (200 g/L potato and 20 g/L sucrose) were inoculated with fungi according to previously described methods. Subsequently, 50 µg/mL kanamycin was placed in 150 mL flasks containing the culture, and the flasks were shaken at 150 rpm at 28 °C for 7 days. Each strain was subjected to this procedure in triplicate. A blood cell counting chamber was used to quantify conidia with three replicates for each treatment.
The fresh dishes B3277, silent transformants, and P1 were inoculated for ferrous iron uptake test. PSA was added with different concentrations of FeSO4 (i.e., –FeSO4, 10 μM FeSO4, and 1.5 mM FeSO4). The colonies were stored under dark conditions and cultured inversely for 14 days. Colony diameter was then measured. Each treatment was repeated three times.
The strains for the pathogenicity assays were initially revitalized from −70 °C, propagated in PS medium for 7 days at 28 °C and shaken at 160 rpm. The concentrations of spores and hyphal suspension were adjusted to 3 × 106 spores/mL in sterilized PS and inoculated in rice panicles at 1 mL per spike before the rice heading stage for 7 days. Each strain was inoculated with ten spikes, and the number of the false smut balls was counted 21 days after the inoculation.
Molecular detection of B3277
PCR was conducted to detect the stability of T-DNA inserted in the B3277 DNA. The mycelium was collected using the method for determining the fungal growth rate. Cetyltrimethylammonium bromide method was used to extract genomic DNA from the mutant B3277 and wild-type P1 strains. We designed G-F/G-R and H-F/H-R primers to detect genes encoding green fluorescent protein (GFP) and hygromycin phosphotransferase (HPH; Table 1) in the T-DNA. The following parameters of the reaction system were used: 2.5 μL of 10 × Trans Taq HiFi PCR buffer (TransGen Biotech Company); 2 μL of dNTPs (2.5 mM); 1 μL of primer (G-F/G-R, H-F/H-R); 0.2 μL of HiFi DNA Polymerase (5 U/μL); 1 μL of genomic DNA; and 16.3 μL of deionized water. Amplification was conducted as follows: initial denaturation at 94 °C for 5 min; followed by 34 cycles of 94 °C denaturation for 30 s; annealing at 60 °C for 30 s; elongation at 72 °C (40 s for GFP and 1.5 min for HPH); and a final extension at 72 °C for 5 min. The PCR products were separated by electrophoresis on an ethidium bromide-stained 1 % agarose gel. The target bands of 636 and 1400 bp length were observed after T-DNA was inserted into the transformants. A gel imaging system was used to obtain images.
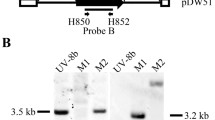
Southern blot analysis results verified the copy number of the transformed T-DNA inserted into the B3277 DNA. Approximately, 5 μg of B3277 and 5 μg of P1 genomic DNA were digested with EcoRI and PstI, respectively, overnight and then separated on 0.8 % agarose gel. DNA was then transferred onto a Hybond N+ membrane. The probe was amplified from the 1.4 kb HPH region using the ALL2 plasmid vector as a template. The probe label, hybridization, and detection were performed according to the manufacturer’s instructions [digoxigenin (DIG) high-prime DNA labeling and detection starter kit I, Roche].
Analysis of the T-DNA flanking sequence of the mutant B3277 strain
The flanking sequence of the B3277 insertion site was obtained through high thermal asymmetric staggered PCR (hiTAIL-PCR) (Liu and Chen 2007). Semi-arbitrary degenerated primers (i.e., LAD5N and LAD6N) and special primers (i.e., AC-L2 and AC-S3) were used. The specific nested primers (i.e., HLB-1, HLB-2, and HLB-3) were designed near the left border (LB) on the HPH gene sequence. HLB-1 and LAD5N in the hiTAIL-PCR were used as pre-amplified primers. The following primers were used in PCR: HLB-2 and AC-S2 (primary) as well as HLB-3 and AC-S3 (secondary) (Fig. 1). The genomic DNA was used as a template. The reaction system and the PCR program were used according to the study (Liu and Chen 2007) with slight modifications. The second round reaction of the hiTAIL-PCR products were cut from the gel, purified, cloned into a pEASY-T3 vector, and transformed into Trans-T1 Escherichia coli cells for sequencing. Sequencing results were then aligned to obtain the B3277 sequence near the LB terminal of the T-DNA. The same approach was used to design the nested primers (i.e., GRB-1, GRB-2, and GRB-3) near the right border (RB) through GFP gene sequencing. The combined primers, particularly GRB-1 and LAD6N, GRB-2 and AC-S2, and GRB-3 and AC-S3, were used in the hiTAIL-PCR.

Schematic diagram of T-DNA insertion event in mutant B3277
Cloning and expression analysis of the mutant gene
The total RNA of the wild-type P1 strain was extracted using a TRIzol reagent kit according to the manufacturer’s instructions. RNA concentration was determined using an ultraviolet spectrophotometer to detect the RNA quality. The PCR reaction system and program were used according to the manufacturer’s instructions of SMARTer RACE cDNA amplification kit. The first strands of 5ʹ-RACE and 3ʹ-RACE were synthesized using the kit according to the manufacturer’s instructions. The primer was designed according to the flanking sequences of the T-DNA insertion site obtained from the hiTAIL-PCR and a longer sequence compared with the genome. Therefore, the first-strand cDNA was used as a template for PCR amplification. The specific primers 5R-1 and 5R-2 were used for 5ʹ-RACE-PCR. 3R-1 and 3R-2 were used for 3ʹ-RACE-PCR (Table 1) based on the obtained pathogenic-related gene fragment. U-Long, U-Short, and 5R-1 were initially used as primers for the touch-down PCR in 5ʹ-RACE-PCR. The PCR product was diluted 100 times as a template for the second round of Nest-PCR using UN-Primer and 5R-2 primers. The PCR product was obtained by gel extraction, cloning, and sequencing. BioEdit was used to combine the gene fragment sequences of 5ʹ-RACE-PCR and 3ʹ-RACE-PCR to obtain the full-length gene.
For real-time PCR (RT-PCR), first-strand cDNA was synthesized using SuperScript III first-strand synthesis system kit. Primers R1 and F1 were used to amplify the first-strand cDNA through PCR. α-Tubulin 2F and α-tubulin 2R (Table 1) were used as reference gene primers. Each reaction was performed in triplicate. The reaction system was used under the following conditions: extension for 20 s and annealing at 60 °C for 30 cycles.
Bioinformatics analysis of the mutant gene
The full-length gene was analyzed by bioinformatics. BlastN alignment (http://www.ncbi.nlm.nih.gov) was searched to determine highly homologous nucleotide sequences. The open reading frame (ORF) of the translated protein was determined using the ORF finder. The amino acids were encoded in NCBI BlastP for sequence and structure analysis. The U. virens Uvt3277 gene encoding the protein function was analyzed using the SMART database (http://smart.embl-heidelberg.de). The DNAMAN 6 sequence alignment software and the neighbor-joining (NJ) MEG4.1 software were used to construct the phylogenetic tree. SignalP-NN was used to predict signal peptides and possible cleavage sites.
Construction of RNAi vector and transformation
An RNAi vector (based on the pSilent-1)was constructed by cloning a fragment (396 bp) of Uvt3277 from the genome of the wild-type P1 using primers S1/S2 and S3/S4 (Nakayashiki et al. 2005) (Fig. S.1). Then the hairpin structure in pSilent1 was subsequently inserted into pCAMBIA 1300 to introduce the silent fragment into U. virens P1 by ATMT.
The selected gene hph was first cloned through PCR to screen RNA-silencing transformants and confirm the introduction of the recombinant vector into P1. RT-PCR was used to test the silencing efficiency of these transformants by cloning their cDNAs. Primers RT-F and RT-R were used for RT-PCR analysis. Transformants with 80 % silencing efficiency were the target silencing transformants.
Results
Enhanced pathogenicity of the mutant B3277
Pathogenicity test was conducted to investigate the incidence of rice false smut 21 days after the rice was inoculated. The results showed the different infection rates of B3277 and P1 in rice (Fig. 2a, b). Approximately, 17–24 and 28–37 diseased grains per spike were found in P1 and B3277, respectively. Therefore, the overall infection rate of B3277 was higher than that in P1. The inserted foreign T-DNA may have affected the negative regulatory gene for pathogenesis, thereby enhancing the virulence of the mutant B3277 strain in rice by reducing or losing the function of the gene.

Comparison of the virulence of P1 and B3277 in the field. a The number of diseased grains caused by B3277 was higher than that of P1. b Statistical analysis of the average diseased grains per spikelet after Liangyoupeijiu was inoculated; two stars indicate that the difference is extremely significant
Molecular test results of B3277
The designed primers of the GFP gene (G-F and G-R) and the HPH (H-F and H-R) were used to amplify the genomic DNA of B3277. The genomic DNA of P1 was used as the control. The target genes of T-DNA could be cloned in the B3277 genome, but not in P1 (Fig. 3a). Southern blot analysis revealed that B3277 contained a single insertion of T-DNA in the genome (Fig. 3b). These results suggested that the T-DNA sequence was stably inserted into the B3277 genome. The inserted sequence was used to clone the related pathogenic genes and analyze the flanking sequence of the T-DNA insertion sites.

Molecular detection of the mutant strain B3277. a PCR detection of GFP and HPH of B3277. Lanes 1 and 2: PCR detection of the GFP genes of P1 and B3277, respectively. Lanes 3 and 4: PCR detection of the HPH of P1 and B3277. b Southern blot detection of B3277, showing that a single T-DNA insertion event occurred in the B3277 genome. Lane 1, genomic DNA of P1 was digested with EcoRI and PstI. Lanes 2 and 3, genomic DNA of B3277 was digested with EcoRI and PstI, respectively. Lane M, DNA marker. c Expression analysis of Uvt3277
Biological characteristics of B3277
The colony of the wild-type strain P1 and mutant B3277 strains were cultured in the PSA, TB3, and MM medium, and then the colony sizes were measured. The colony size in B3277 showed no significant increase in the TB3 medium. However, the average colony sizes of B3277 were 21.22 ± 1.07 and 20 ± 0.88 mm in PSA and MM, respectively, which were significantly lower than that of wild-type strain P1 (22.23 ± 0.38 and 16.70 ± 0.58 mm in PSA and MM cultures, respectively). Moreover, the mycelium of P1 in MM was sparser (Fig. 4a, b). We also measured the sporulation per milliliter. Accordingly, the sporulation quantities were 5.27 ± 0.42 × 106 and 6.92 ± 0.75 × 106 spores/mL in B3277 and P1, respectively (Fig. 4c). The sporulation of the mutant B3277 strain was significantly lower than that in P1.

Comparison of morphology, growth rate, and sporulation of P1 and B3277. a The colony morphology photographed on the front and on the back of P1 and B3277 on PSA, TB3, and MM mediums; B3277 is denser in mycelium, and dark green in PSA and TB3 on the back. b Comparison of the growth rate of B3277 to that of the wild-type P1; B3277 grew slower than P1 in the three types of mediums. One star indicates significant difference. c Comparison of the conidiation of P1 and B3277; the spores in B3277 were fewer than that in P1
The ferrous iron uptake results showed that the mutant strain B3277 could uptake the FeSO4 under certain FeSO4 range concentration. However, no significant influence on growth was shown. By contrast, the P1 growth rate was inhibited with the increasing FeSO4 concentration (Fig. 5).

Influence of ferrous iron uptake on mycelial growth of B3277. a The growth phenotypes of U. virens wild-type P1and mutant strain B3277. b Quantitative analysis of the radial growth of U. virens wild-type P1 and mutant strain B3277 was performed following growth for 14 days at 28 °C
Analysis of the T-DNA insertion sites flanking sequence of B3277
We used specific primers combined with degenerate primers to amplify hiTAIL-PCR and locate the flanking sequences adjacent to the LB terminal of the T-DNA. The length of the obtained sequence of U. virens near the LB was 258 bp, whereas the LB terminal sequence of T-DNA presented in the mutant strain lost 66 bp. The U. virens sequence near the RB terminal of the T-DNA obtained by the same method was 347 bp. However, the RB terminal sequence of the T-DNA segments lost 247 bp.
Cloning and analysis of the full-length gene related to pathogenicity in U. virens
According to the B3277 flanking sequence and the U. virens genome sequencing database, we designed specific primers based on the gene for 5′RACE-PCR and 3′RACE-PCR amplification, cloning, and sequencing. The results showed that the length of the gene was 2946 bp with a 71 bp intron. The 5′ end was extended to 442 bp, and the 3′end was extended to 172 bp. Ultimately, the full-length gene of 3560 bp was accessed. This gene was named as Uvt3277 (GenBank accession no.: KJ158162). RT-PCR was used to analyze Uvt3277 expression and the results showed that the expression was down-regulated in B3277 than in wild strain P1 (Fig. 3c). The T-DNA insertion infected the flanking gene expression, which may be one of the reasons that caused enhanced B3277 pathogenicity.
The results of the bioinformatics analysis of the Uvt3277 gene were presented as follows. The sequence analysis revealed that the gene comprised two exons with lengths of 890 and 2670 bp and an intron with 71 bp, coding a protein with 517 amino acids. The T-DNA insertion site was in the middle of the second exon. The intron shear laws conformed to GT/AG. The full-length cDNA sequence of the gene was analyzed by the NCBI ORF finder. The gene has 1554 bp of the complete ORF with an 1158 bp length of 5’ untranslated regions (5′UTR) and an 848 bp length of 3′UTR. The gene Uvt3277 was homologous to Uv8b_7257 in U. virens Uv8b (Zhang et al. 2014). However, the ORF of Uvt3277 was 448 bp shorter than that of Uv8b_7257. Uvt3277 only had a 71 intron in 5′UTR, while Uv8b_7257 had the same intron and another 80 bp intron in ORF (Fig. S.2).
Comparative analysis of the full-length gene sequence from the NCBI BlastN indicated that Uvt3277 was highly similar (reaching 73 %) to the low-affinity iron transporter Aspergillus oryzae RIB40 mRNA (XM_001825832.2). Both NCBI BlastP and SMART database analyses indicated that the protein contains two small integral membrane proteins and three low-affinity iron permeases.
Proteins exhibiting high similarity to the protein encoded by Uvt3277 were searched in GenBank. A large number of homologous protein sequences were found in fungi with low-affinity iron transport. The top six scoring sequences were Metarhizium acridum CQMa 102, Metarhizium anisopliae ARSEF 23, Beauveria bassiana ARSEF 2860, Cordyceps militaris CM01, Aspergillus fumigatus A1163, and Aspergillus oryzae RIB40. Phylogenetic tree analysis showed that the proteins of B3277, CQMa 102, and ARSEF 23 were clustered on the same branch (Fig. 6). The primary protein structure of Uvt3277 was then analyzed. The measured theoretical molecular mass of the protein is 129.639 kDa, and the predicted isoelectric point (pI) is 4.9. The unstable factor of the unstable protein is 55.84. The average pI of the hydrophilic residue is 0.921. Positively or negatively charged residues, including Ala, Cys, Gly, and Thr, yielded an average pI of 0. The SignalP-NN analysis revealed no signal peptide in the polypeptide chain.

Amino acid sequence alignment and phylogenetic analysis of Uvt3277 protein with homologs from other fungi. Black highlight indicates that the amino acids were identical in all of the segments, and red highlight indicates that six of the seven amino acids are identical at the indicated positions. All of the amino acid sequences of Uvt3277 homologs except the Uvt3277 protein derived from this study are obtained from GenBank. These sequences include Metarhizium acridum CQMa 102 (EFY91699.1), Metarhizium anisopliae ARSEF 23 (EFY94670.1), Beauveria bassiana ARSEF 2860 (EJP70819.1), Cordyceps militaris CM01 (XP_006669511.1), Aspergillus fumigatus A1163 (EDP50848.1), and Aspergillus oryzae RIB40 (XP_001825884.1). The numbers at the branch nodes are bootstrap values
Construction of RNAi vector and transformation
The RNA silencing mechanism was exploited as a novel and efficient genetic tool for gene function analysis using an hpRNA-expressing plasmid or an opposing dual promoter system in filamentous fungi. The fungal protoplast or conidium treatment with small-interfering RNAs (siRNAs) could reduce the expression of their target genes. pSilent1-Uvt3277 was constructed by inserting PCR products into multiple cloning sites (Fig.S.3) and subsequently introducing it into the wild-type strain P1 to generate hpRNA of the Uvt3277 fragment (396 bp).
The expression levels of Uvt3277 in Uvt327-silencing transformants were decreased to 10–90 % compared with those in P1 (Fig. 7a). The phenotypes of six representative transformants with different silencing efficiency were studied. Three (S21, S19, S9) have 10–30 % levels of Uvt3277 mRNA compared to the wild-type strain. The levels were about 50 % in transformants S4 and 70–90 % in transformants S5 and S16. The six transformants were checked for phenotypes in growth, sporulation, and the ability of uptaking FeSO4. There was a strong correlation between the levels of silencing and the abnormalities. For example, transformant S21 exhibited the highest decrease of the mRNA level and showed the abnormalities consistent with B3277 (Fig. 7b, c, d). The S21 pathogenicity was also enhanced compared with the wild-type strain P1 (Fig. 8a, b), which was similar to the strain B3277 (the mRNA level was about 85 % decreased). Approximately, 10–32 and 14–78 diseased grains per spike were found in P1 and S21, respectively. These phenotypes confirmed that Uvt3277 had an important function in pathogenesis.

a The growth phenotypes of U. virens wild-type P1 and mutant strain S21 under ferrous iron uptake. b The growth phenotypes of U. virens wild-type P1 and mutant strain S21 under ferrous iron uptake. c Quantitative analysis of the radial growth of U. virens wild-type P1 and mutant strain S21 was performed following growth for 14 days at 28 °C. d Comparison of the conidiation of P1 and S21; the spores in S21 are fewer than those in P1

Comparison of the pathogenicity of P1 and S21 in the field. a Statistical analysis of the average diseased grains per spikelet after Liangyoupeijiu was inoculated; two stars indicate that the difference is extremely significant. b The number of diseased grains caused by S21 was much higher than that by P1
Discussion
The T-DNA induces the expression of certain genes. Moreover, the T-DNA as the insertion element can damage the gene function in the insertion site and may change the phenotype and biochemical characteristics of the mutant strains. (Bourras et al. 2012; Singer et al. 2012). The genetic transformation mediated by the ATMT has been extensively performed to study the function of the pathogenicity genes in some fungi (Maruthachalam et al. 2012). This study selects a single T-DNA insertion mutant with enhanced virulence (i.e., B3277) to investigate the function of the pathogenicity genes in U. virens. The biological characteristics of the mutant B3277 strain indicated that the growth rate and sporulation of B3277 were decreased compared with those of the wild-type strain P1. However, the virulence of B3277 was significantly increased. The T-DNA was inserted in the middle of the gene, and RT-PCR results revealed that the expression level of Uvt3277 was decreased. Therefore, we speculated that Uvt3277 was a negative regulatory factor associated with pathogenicity.
Li et al. (2007) reported a number of RB and LB flanking sequences of the T-DNA insertion mutants in M.oryzae. The LB of the T-DNA is inaccurately nicked and frequently truncated during the integration compared with RB. In here, the flanking sequences of T-DNA in B3277 were analyzed. A 231 bp LB flanking sequence is found, which reveals that 66 bp has been lost. However, the RB has almost lost the entire sequence length, leaving only 55 bp from the original 302 bp. Therefore, the LB is more conserved than the RB. This result is different from that in other studies (Fu et al. 2006; Kemski et al. 2013; Li et al. 2007). Considering that the T-DNA precisely and easily integrates into filamentous fungi, we assume that this behavior may be an accidental pattern exhibited by U. virens.
The Uvt3277 protein is aligned with the homologous sequence and shares a high homology with low-affinity iron transport protein in some fungi. Uvt3277 possesses the typical ferroportin functional structure that contains two small integral membrane proteins and three low-affinity iron permeases, which is the complete functional domain of the Fe family. Therefore, this gene is likely involved in the function of ferrous ion transporters. Fe is very important for plants to develop normally. This element is also involved in many biological functions. Iron deficiency or excess could cause harmful effects to plants (Li and Chen. 2013). The transport proteins sometimes function as a secondary regulator that influences gene expression and metabolism (Västermark and Saier 2014). Fe homeostasis is the crucial role in plant pathogen interactions and in particular defense responses. Modulating Fe supply leads to different outcomes, depending on the pathogen infection strategy (Aznar et al. 2015). Yeasts can be considered as efficient model organisms in studies involving the iron transport mechanism. For example, Saccharomyces cerevisiae uses four pathways to accumulate iron. These pathways include siderophore iron accumulation, high-affinity iron uptake, and two low-affinity uptake pathways via divalent metal ion transporters (Kosman 2003). The low-affinity transporter system contains products of the SMF1 and FET4 genes (Kaplan et al. 2006). The high-affinity iron uptake systems are not expressed. Iron uptake also occurs via low-affinity transporters exhibiting low specificity to divalent metal transporters (e.g., Fet4p, Smf1p, Smf2p, and Smf3p) when iron is freely available to yeasts (Philpott 2006). Two major virulence factors are found in the pathogenic fungus Cryptococcus neoformans; polysaccharide capsule and melanin induce pathogens to compete with hosts and acquire iron, which can regulate the expression of virulence factors that cause infection (Jung and Kronstad 2008). Fungi evolve and develop efficient mechanisms for iron uptake and storage. Two major transcription factors, GATA-factor SreA and bZip-factor HapX, are regulated in A. fumigatus by iron levels. Moreover, iron is a key nexus to determine virulence (Haas 2012; Schrettl and Haas 2011). The relative machinery of these transport proteins has been related to disease (Jose et al. 2013). Ustilaginoidea virens is a biotrophic fungal pathogen and can uptake iron from rice which may be helpful for the synthesis of virulence factors. The Uvt3277 gene in this study is expressed at a very low level. However, virulence is enhanced. We speculate that there may be several pathways for U. virens to accumulate iron from the host. Uvt3277 is a low-affinity iron transport protein. If the gene is broken and the pathway does not work, U. virens may choose another high-affinity iron uptake pathway to get more iron; thus, the rice cannot grow normally and more virulence factors of U. virens could be synthesized, causing enhanced pathogenicity. Therefore, iron may have important functions in U. virens pathogenesis and Uvt3277 is a key gene related to the low-affinity iron transport.
Further studies were conducted regarding knockdown of the gene Uvt3277 expression by RNA interference (RNAi). The RNAi plays a critical role in gene regulation in various eukaryotic organisms. RNA silencing or RNAi is a gene regulatory system used through double-stranded RNA (dsRNA)-mediated homology-dependent gene inactivation in eukaryotic organisms. In this process, dsRNA that can be induced by hairpin RNA (hpRNA)-induced gene silencing encoded RNA (Waterhouse and Helliwell 2003) is degraded into siRNAs. These siRNAs are incorporated into the RNA-induced silencing complex to target and degrade the gene mRNA with the complementary sequence (Liu et al. 2002; McDonald et al. 2005; Nakayashiki and Nguyen 2008; Waterhouse and Helliwell 2003). RNAi silences genes in a number of fungi (Kück and Hoff 2010) and examples include C. neoformans (Liu et al. 2002), M. oryzae (Jeon et al. 2007; Kadotani et al. 2003; Nakayashiki et al. 2005), and Aspergillus and Fusarium species (McDonald et al. 2005). The pSilent1-Uvt3277 knockdown carrier was constructed and inserted into P1 using the ATMT method. The mutant S21 was assessed to determine the relationship between the gene and its function in pathogenesis. This study may provide a theoretical basis for Uvt3277 in the pathogenic process of U. virens and new prevention and treatment of rice false smut.
In conclusion, T-DNA insertion affects the expression of Uvt3277 and consequently increased U. virens pathogenicity. The full length of the destroyed gene is cloned and analyzed. Uvt3277, which is a low-affinity iron transport protein, verifies the relationship with pathogenicity by RNAi. Further studies will focus on the interacting proteins of Uvt3277 in U. virens and explore insights into the pathogenesis mechanism.
References
Andargie M, Li J (2016) Arabidopsis thaliana: a model host plant to study plant–pathogen interaction using rice false smut isolates of Ustilaginoidea virens. Front Plant Sci 7:192
Ashizawa T, Takahashi M, Moriwaki J, Hirayae K (2010) Quantification of the rice false smut pathogen Ustilaginoidea virens from soil in Japan using real-time PCR. Eur J Plant Pathol 128:221–232
Ashizawa T, Takahashi M, Moriwaki J, Hirayae K (2011) A refined inoculation method to evaluate false smut resistance in rice. J Gen Plant Pathol 77:10–16
Ashizawa T, Takahashi M, Arai M, Arie T (2012) Rice false smut pathogen, Ustilaginoidea virens, invades through small gap at the apex of a rice spikelet before heading. J Gen Plant Pathol 78:255–259
Aznar A, Chen NW, Thomine S, Dellagi A (2015) Immunity to plant pathogens and iron homeostasis. Plant Sci 240:90–97
Bourras S, Meyer M, Grandaubert J, Nicolas L, Isabelle F, Juliette L, Benedicte O, Françoise B, Marie-Hélène B, Thierry R (2012) Incidence of genome structure, DNA asymmetry, and cell physiology on T-DNA integration in chromosomes of the phytopathogenic fungus Leptosphaeria maculans. G3 Genes Genomes Genetics 2:891–904
Chi M-H, Park S-Y, Kim S, Lee Y-H (2009) A novel pathogenicity gene is required in the rice blast fungus to suppress the basal defenses of the host. PLoS Pathogol 5:e1000401
de Groot MJ, Bundock P, Hooykaas PJ, Beijersbergen AG (1998) Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nature Biotechnology 16
Ebbole DJ (2007) Magnaporthe as a model for understanding host-pathogen interactions. Annu Rev Phytopathol 45:437–456
Fan YQ, Zhang S, Yu DZ, Gong Y (2012) Research progress on Ustilaginoidea virens. HuBei Agric Sci 21:4701–4704
Fang A, Han Y, Zhang N, Zhang M, Liu L, Li S, Lu F, Sun W (2016) Identification and characterization of plant cell death–inducing secreted proteins from Ustilaginoidea virens. Mol Plant Microbe Interact 29(5):405–416
Fu D, St Amand PC, Xiao Y, Muthukrishnan S, Liang GH (2006) Characterization of T-DNA integration in creeping bentgrass. Plant Sci 170:225–237
Guo X, Li Y, Fan J, Li L, Huang F, Wang W (2012) Progress in the study of false smut disease in rice. J Agric Sci Technol A 11:1211–1217
Haas H (2012) Iron–a key nexus in the virulence of Aspergillus fumigatus. Front Microbiol 3
Hu J, Chen Z, Gu CH, Yu M, Yu J, Nie Y, Huang L, Qiao J, Huang X, He J, Liu Y (2013) Isolation and characterization of a promoter from rice false smut fungus Ustilaginoidea virens. J Plant Pathol 95(3):539–547
Hu M, Luo L, Wang S, Liu Y, Li J (2014) Infection processes of Ustilaginoidea virens during artificial inoculation of rice panicles. Euro J Plant Pathol 139:67–77
Jeon J, Park S, Chi M, Choi J, Park J, Rho H, Kim S, Goh J, Yoo S, Choi J, Park J, Yi M, Yang S, Kwon M, Han S, Kim B, Khang C, Park B, Lim S, Jung K, Kong S, Karunakaran M, Oh H, Kim H, Kim S, Park J, Kang S, Choi W, Kang S, Lee Y (2007) Genome-wide functional analysis of pathogenicity genes in the rice blast fungus. Nat Genet 39:561–565
Jiang D, Zhu W, Wang Y, Sun C, Zhang K-Q, Yang J (2013) Molecular tools for functional genomics in filamentous fungi: recent advances and new strategies. Biotechnol Adv 31:1562–1574
Jose MM, Vicent LT, Cecilia P, Ma CM, Lynne Y (2013) Endocytic regulation of alkali metal transport proteins in mammals, yeast and plants. Curr Genet 59:207–230
Jung WH, Kronstad JW (2008) Iron and fungal pathogenesis: a case study with Cryptococcus neoformans. Cell Microbiol 10:277–284
Kadotani N, Nakayashiki H, Tosa Y, Mayama S (2003) RNA silencing in the phytopathogenic fungus Magnaporthe oryzae. Mol Plant Microbe Interact 16:769–776
Kaplan J, McVey Ward D, Crisp RJ, Philpott CC (2006) Iron-dependent metabolic remodeling in S. cerevisiae. Biochim Biophys Acta Mol Cell Res 1763:646–651
Kemski MM, Stevens B, Rappleye CA (2013) Spectrum of T-DNA integrations for insertional mutagenesis of Histoplasma capsulatum. Fungal Biol 117:41–51
Kosman DJ (2003) Molecular mechanisms of iron uptake in fungi. Mol Microbiol 47:1185–1197
Kück U, Hoff B (2010) New tools for the genetic manipulation of filamentous fungi. Appl Microbiol Biotechnol 86:51–62
Li S-Z, Chen J-T (2013) Progresses in studying of protein families involved in Zn/Fe transporting in plants. Biotechnol Bull 2:8–14
Li G, Zhou Z, Liu G, Zheng F, He C (2007) Characterization of T-DNA insertion patterns in the genome of rice blast fungus Magnaporthe oryzae. Curr Genet 51:233–243
Li Y, Liang S, Yan X, Wang H, Li D, Soanes DM, Talbot NJ, Wang Z, Wang Z (2010) Characterization of MoLDB1 required for vegetative growth, infection-related morphogenesis, and pathogenicity in the rice blast fungus Magnaporthe oryzae. Mol Plant Microbe Interact 23:1260–1274
Liu Y-G, Chen Y (2007) High-efficiency thermal asymmetric interlaced PCR for amplification of unknown flanking sequences. Biotechniques 43:649–656
Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering TL (2002) RNA interference in the pathogenic fungus Cryptococcus neoformans. Genetics 160:463–470
Maruthachalam K, Klosterman S, Kang S, Hayes R, Subbarao K (2011) Identification of pathogenicity-related genes in the vascular wilt fungus Verticillium dahliae by Agrobacterium tumefaciens-mediated T-DNA insertional mutagenesis. Mol Biotechnol 49:209–221
Maruthachalam K, Jeon J, Lee Y-H, Subbarao KV (2012) Identification of fungal pathogenicity genes by Agrobacterium tumefaciens-mediated transformation. Biotech Fungal Genes:1
McDonald T, Brown D, Keller NP, Hammond TM (2005) RNA silencing of mycotoxin production in Aspergillus and Fusarium species. Mol Plant Microbe Interact 18:539–545
Meng J, Sun W, Mao Z, Xu D, Wang X, Lu S, Lai D, Liu Y, Zhou L, Zhang G (2015) Main ustilaginoidins and their distribution in rice false smut balls. Toxins 7(10):4023–4034
Michielse CB, Hooykaas PJ, van den Hondel CA, Ram AF (2005) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48:1–17
Michielse CB, van Wijk R, Reijnen L, Cornelissen B, Rep M (2009) Insight into the molecular requirements for pathogenicity of Fusarium oxysporum f. sp. lycopersici through large-scale insertional mutagenesis. Genome Biol 10:R4
Mullins E, Chen X, Romaine P, Raina R, Geiser D, Kang S (2001) Agrbacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180
Nakayashiki H, Nguyen QB (2008) RNA interference: roles in fungal biology. Curr Opin Microbiol 11:494–502
Nakayashiki H, Hanada S, Quoc NB, Kadotani N, Tosa Y, Mayama S (2005) RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet Biol 42:275–283
Philpott CC (2006) Iron uptake in fungi: a system for every source. Biochim Biophys Acta Mol Cell Res 1763:636–645
Schrettl M, Haas H (2011) Iron homeostasis—Achilles’ heel of Aspergillus fumigatus. Curr Opin Microbiol 14:400–405
Singer K, Shiboleth YM, Li J, Tzfira T (2012) Formation of complex extrachromosomal T-DNA structures in Agrobacterium tumefaciens-infected plants. Plant Physiol 160:511–522
Sun X, Kang S, Zhang Y, Tan X, Yu Y, He H, Zhang X, Liu Y, Wang S, Sun W, Cai L, Li S (2013) Genetic diversity and population structure of rice pathogen Ustilaginoidea virens in China. PLoS One 8:e76879
Talbot NJ (2003) On the trail of a cereal killer: exploring the biology of Magnaporthe grisea. Ann Rev Microbiol 57:177–202
Tanaka E, Ashizawa T, Sonoda R, Tanaka C (2008) Villosiclava virens gen. nov., comb. nov., the teleomorph of Ustilaginoidea virens, the causal agent of rice false smut. Mycotaxon 106:491–501
Tang Y, Jin J, Hu D, Yong M, Xu Y, He L (2013) Elucidation of the infection process of Ustilaginoidea virens (teleomorph: Villosiclava virens) in rice spikelets. Plant Pathol 62:1–8
Valent B (1990) Rice blast as a model system for plant pathology. Phytopathology 80:33–36
Västermark Å, Saier MH (2014) The involvement of transport proteins in transcriptional and metabolic regulation. Curr Opin Microbiol 18:8–15
Waterhouse PM, Helliwell CA (2003) Exploring plant genomes by RNA-induced gene silencing. Nat Rev Genet 4:29–38
White Jr JF, Sullivan R, Moy M, Patel R, Duncan R (2000) An overview of problems in the classification of plant-parasitic Clavicipitaceae. Studies in Mycology 45(45):95–105
Yin X, Chen Z, Liu Y, Yu J, Li Y, Yu M (2012) Detection of the relative content of ustiloxin A in rice false smut balls and correlation analysis between pathogenicity and ustiloxins A production of Ustilaginoidea virens. Sci Agric Sin 45:4720–4727
Yu M, Yu J, Hu J, Huang L, Wang Y, Yin X, Nie Y, Meng X, Wang W, Liu Y (2015) Identification of pathogenicity-related genes in the rice pathogen Ustilaginoidea virens through random insertional mutagenesis. Fungal Genet Biol 76:10–19
Zhang Y, Zhang K, Fang A, Han Y, Yang J, Xue M, Bao J, Hu D, Zhou B, Sun X, Li S, Wen M, Yao N, Ma L, Liu Y, Zhang M, Huang F, Luo C, Zhou L, Li J, Chen Z, Miao J, Wang S, Lai J, Xu J, Hsiang T, Peng Y, Sun W (2014) Specific adaptation of Ustilaginoidea virens in occupying host florets revealed by comparative and functional genomics. Nat Commn 5:3849–3860
Zhou YL, Izumitsu K, Sonoda R, Nakazaki T, Tanaka E, Tsuda M, Tanaka C (2003) PCR-based specific detection of Ustilaginoidea virens and Ephelis japonica. J Phytopathol 151:513–518
Zhou Y, Pan Y, Xie X, Zhu L, Xu J, Wang S, Li Z (2008) Genetic diversity of rice false smut fungus, Ustilaginoidea virens and its pronounced differentiation of populations in North China. J Phytopathol 156:559–564
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31401700), the Agriculture Innovation Foundation of Jiangsu Province (CX(12)5005, CX(15)1054).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Communicated by M. Kupiec.
Meng-ting Zheng and Hui Ding are co-first authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, Mt., Ding, H., Huang, L. et al. Low-affinity iron transport protein Uvt3277 is important for pathogenesis in the rice false smut fungus Ustilaginoidea virens . Curr Genet 63, 131–144 (2017). https://doi.org/10.1007/s00294-016-0620-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0620-4