
Abstract
Chaperones of the diverse ubiquitous Hsp70 family are involved in the regulation of ordered self-perpetuating protein aggregates (amyloids and prions), implicated in both devastating diseases and protein-based inheritance. Yeast ribosome-associated chaperone complex (RAC), composed of the Hsp40 protein Zuo1 and non-canonical Hsp70 protein Ssz1, mediates association of the Hsp70 chaperone Ssb with translating ribosomes. Ssb participates in co-translational protein folding, regulation of premature translation termination, and ribosome biogenesis. The loss of Ssb or disruption of RAC results in the increased formation of [PSI +], a prion form of the translation termination factor Sup35 (eRF3). This implicates co-translational protein misfolding in de novo prion formation. However, RAC disruption also destabilizes pre-existing [PSI +] prions, as Ssb, released from ribosomes to the cytosol in the absence of RAC, antagonizes the function of the major cytosolic chaperone, Ssa, in prion propagation. The mechanism of the Ssa/Ssb antagonism is currently under investigation and may include a competition for substrates and/or co-chaperones. Notably, yeast cells with wild-type RAC also release Ssb to the cytosol in certain unfavorable growth conditions, and Ssb contributes to increased prion loss in these conditions. This indicates that the circulation of Ssb between the ribosome and cytosol may serve as a physiological regulator of the formation and propagation of self-perpetuating protein aggregates. Indeed, RAC and Ssb modulate toxicity of some aggregating proteins in yeast. Mammalian cells lack the Ssb ortholog but contain a RAC counterpart, apparently recruiting other Hsp70 protein(s). Thus, amyloid modulation by ribosome-associated chaperones could be applicable beyond yeast.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Hsp70/DnaK chaperones are evolutionarily conserved proteins that are found from bacteria to humans and play a key role in a variety of protein folding processes, as well as in protection from stress. In yeast Saccharomyces cerevisiae, a model organism frequently employed in genetic and biochemical studies, the major cytoplasmic subfamilies of the Hsp70 family include the evolutionarily conserved Ssa subfamily, coded by 4 genes, SSA1-4, and the fungal-specific Ssb subfamily, coded by 2 almost identical genes, SSB1 and SSB2 (for review, see Reidy and Masison 2011; Rikhvanov et al. 2007; Sharma and Masison 2009). The Ssa subfamily consists of both constitutive and stress-inducible proteins, and the presence of at least one Ssa protein is essential for cell viability (Werner-Washburne et al. 1987; Werner-Washburne and Craig 1989). Ssa chaperones are aided by the Hsp40 proteins Sis1 or Ydj1, and participate in a variety of posttranslational protein folding events, as well as (together with chaperone Hsp104) in disaggregation and refolding of proteins damaged by stress (Reidy and Masison 2011; Rikhvanov et al. 2007). In contrast, Ssb is not essential, is not induced by heat shock, and is associated with translating ribosomes. This association is mediated by the Hsp40 co-chaperone Zuo1 and non-conventional Hsp70-derived co-chaperone Ssz1, together constituting the ribosome-associated chaperone complex, RAC (Fiaux et al. 2010; Gautschi et al. 2001; Huang et al. 2005). RAC and Ssb have been shown to participate in co-translational folding of nascent polypeptides (Fiaux et al. 2010; Gautschi et al. 2001; Huang et al. 2005; James et al. 1997; Nelson et al. 1992). Coupling of co-translational folding with peptide elongation (Zhang et al. 2014) has been demonstrated to modulate premature termination of translation on the unusual mRNA regions causing ribosome stalling, such as in the case of polylysines (Chiabudini et al. 2012, 2014). Zuo1 and Ssb also play a role in ribosome biogenesis (Albanese et al. 2010). In addition, Zuo1 and Ssz1 proteins are implicated in the modulation of drug resistance and quorum sensing via regulation of a membrane transporter (Prunuske et al. 2012). RAC dissociation as a result of deletion of ZUO1 and/or SSZ1 leads to release of Ssb from the ribosome to the cytosol (Willmund et al. 2013). Zuo1/Ssz1 deficiency or Ssb depletion leads to accumulation of misfolded aggregated proteins in the yeast cell (Koplin et al. 2010; Willmund et al. 2013). This set of proteins is enriched in longer and slowly translated polypeptides that possess intrinsically disordered regions, are characterized by increased hydrophobic exposure and aggregation potential, and have a higher propensity for forming β-structures.
Aggregation of β-sheet rich proteins is known to produce ordered self-perpetuating protein polymers (amyloids). A variety of proteins from various organisms, encompassing the whole evolutionary spectrum from bacteria to humans, have been shown to form amyloids (Aguilar-Calvo et al. 2015; Blanco et al. 2012; Buxbaum and Linke 2012; Fowler et al. 2007; Wickner et al. 2015). Amyloids and their transmissible (infectious or heritable) forms termed prions are associated with devastating mammalian and human diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s diseases, and transmissible spongiform encephalopathies. However, some amyloids also play biologically positive roles. Endogenous transmissible amyloids in yeast (yeast prions) were shown to be associated with pathogenic processes, but are also implicated in some potentially adaptive functions. Yeast prions manifest themselves as non-Mendelian elements that control cytoplasmically inherited traits. About 10 amyloid-based yeast prions are described to date (for review, see Chernova et al. 2014; Liebman and Chernoff 2012). Among them, the prion [PSI +], formed by the translation termination factor Sup35 (eRF3), is arguably the best studied. [PSI +] formation leads to a defect in termination of translation, which provides a convenient phenotypic assay for [PSI +] based on the readthrough of nonsense mutations (nonsense-suppression). This phenotype is easily detectable by growth or color of specially designed strains on certain media (see Liebman and Chernoff 2012) and similar to the phenotype caused by mutations in the SUP35 gene (see Nizhnikov et al. 2014; Protacio et al. 2015). Propagation of almost all known yeast prions depends on the chaperone machinery, which is composed of the AAA + (ATPases associated with a variety of cellular activities) chaperone Hsp104 and cytosolic members of the Hsp70 (Ssa) and Hsp40 (Ydj1 or Sis1) families (for review, see Chernova et al. 2014; Liebman and Chernoff 2012; Reidy and Masison 2011). This chaperone machinery promotes fragmentation of prion polymers into oligomeric seeds (“propagons”), thus initiating new rounds of prion propagation (Fig. 1a), which is required for the “vertical” prion transmission to daughter cells and, potentially, for the “horizontal” transmission between neighbor cells via extracellular vesicles (Kabani and Melki 2015a, b). A lack or decreased activity of Hsp104 results in a polymer fragmentation defect and segregational loss of prion polymers in cell divisions (Fig. 1b). When present in excess of Ssa, Hsp104 may bind some prion aggregates on its own, however, it cannot generate propagons (Fig. 1c). This leads to destabilization and loss of the [PSI +] prion, although most other prions remain unaffected (Chernova et al. 2014; Liebman and Chernoff 2012). Ribosome-associated chaperones, participating in folding of aggregation-prone nascent polypeptides, are naturally positioned to regulate prion formation and abundance. However, the scope of impact of ribosome-associated chaperones on prions was poorly understood till now. This paper, written as an extension of our recent experimental work (Kiktev et al. 2015), reviews data from our and other labs to decipher the impact of ribosome-associated chaperones on the formation and propagation of a yeast prion.

Role of Hsp104 and Hsp70 in the propagation of [PSI +] prion. a Cooperative binding of Ssa (assisted by Hsp40 cochaperones, not shown on figure) and Hsp104 assures proper fragmentation of prion fibers into smaller oligomers (propagons), which act as “seeds” for new rounds of prion propagating. b Decrease in the Hsp104/Ssa ratio results in prion loss via asymmetric segregation in the absence of fragmentation. c Increase in the Hsp104/Ssa ratio results in “non-productive” Hsp104 binding and subsequent prion loss via asymmetric segregation in the absence of fragmentation, or “chopping” monomers from the fiber ends, or a combination of both. Dark squares units of prion polymer, dark circle non-prion monomeric Sup35, striped circle Ssa1, hexagon Hsp104
Differential effects of Ssa and Ssb chaperones on the [PSI +] prion
The current model states that the Hsp104/Ssa/Sis1 (or Ydj1) machinery is responsible for the fragmentation of prion aggregates initiating new rounds of prion propagation (for review, see Chernova et al. 2014; Liebman and Chernoff 2012). When the Hsp104/Ssa ratio is impaired by either decreasing or increasing the Hsp104 levels or activity, the [PSI +] prion is destabilized (Fig. 1). Notably, simultaneous overproduction of Ssa partly restores [PSI +] propagation in the presence of high levels of Hsp104 (Newnam et al. 1999). Previously, we have shown that, in contrast to Ssa, the overproduction of Ssb facilitates [PSI +] elimination by excess Hsp104, while double deletion of the SSB1 and SSB2 genes (ssb1/2∆) ameliorates [PSI +] loss in these conditions (Chernoff et al. 1999). Continuous overproduction of Ssb also destabilizes some variants of [PSI +] prion (Chacinska et al. 2001; Kushnirov et al. 2000). Moreover, de novo [PSI +] formation is significantly increased in the ssb1/2∆ strain, implicating ssb1/2∆ as a “protein mutator”, increasing the frequency of heritable conformational change in another protein (Chernoff et al. 1999). Each Hsp70 protein consists of three major regions (Fig. 2): N-terminal nucleotide-binding domain (NBD), substrate- (or peptide-) binding domain (SBD), and C-terminal variable domain (CTD, or “lid”) (Rikhvanov et al. 2007; Sharma and Masison 2009). Our experiments with chimeric constructs pointed to SBD as the major determinant of differences in the effects of Ssa and Ssb on a prion (Allen et al. 2005).

Structural and functional organization of yeast Hsp70 proteins from Ssa (Ssa1) and Ssb (Ssb1) subfamilies. N-terminal nucleotide binding (NBD), substrate binding (SBD), and C-terminal (CTD) are shown. Numbers represent amino acid positions used to swap domains of these two proteins (James et al. 1997) to produce Ssa/Ssb chimeric constructs employed later in this work (see below, Fig. 4b–e and related text)
Role of ribosome-associated chaperone complex (RAC) in de novo prion formation
Recently, we (Kiktev et al. 2015) and others (Amor et al. 2015) demonstrated that deletion of either ZUO1 or SSZ1 increases both spontaneous de novo [PSI +] formation and induction of [PSI +] formation by transient overproduction of the Sup35 prion domain (PrD) in a manner similar to ssb1/2Δ. This effect is more pronounced in the presence of Rnq1 prion, [PIN +], which is known to facilitate de novo formation of [PSI +] (Derkatch et al. 1997, 2001; Osherovich and Weissman 2001). However, in certain experimental designs, an increase in [PSI +] formation could also be detected in RAC-deficient cells lacking the Rnq1 prion. The effects of ssb1/2Δ and triple deletions (ssb1/2Δ zuo1Δ and ssb1/2Δ ssz1Δ) on [PSI +] formation were identical to each other, indicating that all of these proteins influence prion induction via the same pathway. Increased [PSI +] formation can be explained by accumulation of misfolded proteins, potentially convertible into a prion, on the ribosomes lacking Ssb (Fig. 3a, b), and is consistent with the role of Ssb-RAC in the folding and quality control of nascent polypeptides.

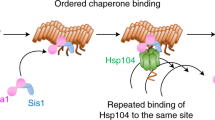
Dual role of RAC-Ssb in the formation and propagation of [PSI +] prion. a In wild-type (WT) cells, most Ssb (solid white circle) is bound to translating ribosomes via the RAC complex, composed of Zuo1 and Ssz1 proteins (sectored white circle). This facilitates proper folding of the nascent polypeptide into a non-prion monomer (solid dark circle), and therefore prevents de novo prion formation. Pre-existing prion polymers are propagated via the Hsp104/Ssa machinery (see Fig. 1 for details and designations), with the help of cofactors such as Hsp40 cochaperones and nucleotide exchange factors (dark triangle). b In cells lacking one or both RAC components, or in the cells incubated in poor medium, all or most Ssb is released from the ribosomes to cytosol. This increases misfolding of the nascent polypeptide, leading to de novo prion formation. However, cytosolic Ssb interferes with Ssa function, impairing [PSI +] propagation and facilitating non-productive binding of Hsp104 that results in prion loss. Three possible mechanisms of Ssb interference with Ssa are indicated: direct competition between Ssb and Ssa for binding to prion polymers (left); sequestration of Ssa cofactors by Ssb that prevents Ssa binding to prion polymers (center); formation of the Ssb/Ssa heterodimer that is incapable of prion binding and/or nonfunctional in Hsp104 binding (right)
Effects of the RAC disruption on prion propagation
Unexpectedly, our data have shown that the Ssb-RAC disassembly interferes with prion propagation in a manner similar to the effect of Ssb overproduction rather than to the effect of ssb1/2Δ (Kiktev et al. 2015). Deletion of ZUO1 and/or SSZ1 facilitated [PSI +] elimination by excess Hsp104, and increased spontaneous loss of the weak [PSI +] variants even at normal levels of Hsp104. Moreover, the “anti-prion” effect of zuo1Δ or ssz1Δ was eliminated in combination with ssb1/2Δ, indicating that it is mediated by Ssb rather than resulting from the accumulation of misfolded proteins due to Ssb-RAC disassembly. In agreement with this notion, Ssb overproduction, known to antagonize weak [PSI +] variants (Chacinska et al. 2001; Kushnirov et al. 2000), further increased [PSI +] loss in the zuo1Δ or ssz1Δ strains. The size of Sup35 prion polymers was somewhat increased in RAC-deficient cells compared to isogenic wild type cells, suggestive of a prion fragmentation defect (Kiktev et al. 2015). Levels of Sup35, Ssb, Hsp104, Ssa and other major chaperones were not altered in the zuo1Δ or ssz1Δ strains, ruling out the possibility of an indirect effect on [PSI +] via induction of stress response. However, the anti-[PSI +] effects of RAC dissociation and/or Ssb overproduction were partly suppressed by overproduction of Ssa1. Overall, these data show that the release of Ssb from the ribosome to the cytosol in cells with RAC disruptions counteracts the propagation of pre-existing [PSI +] prion by antagonizing an essential component of the prion fragmentation machinery, Ssa (Fig. 3b).
Physiological role of prion regulation by RAC and Ssb
Taken together, our data show that Ssb-RAC dissociation increases de novo prion formation, probably due to increased protein misfolding in the absence of Ssb on the ribosome. However, Ssb-RAC dissociation also antagonizes propagation of prion aggregates generated in this process, apparently because cytosolic Ssb antagonizes the binding of Ssa to prion aggregates. This generates a regulatory circuit in which Ssb released from ribosomes to the cytosol can interfere with the propagation of prion aggregates that are generated as a result of its release. Is this circuit an artifact of mutational RAC disruption, or does it reflect a physiological mechanism regulating protein aggregation in yeast cells? We have found that Ssb is partially released into cytosol in wild-type cells growing in certain conditions, for example in poor synthetic medium, SC (Kiktev et al. 2015). Notably, spontaneous [PSI +] loss was increased at more than 104-fold in the wild-type strain continuously growing in SC, compared to complete organic (YPD) medium. While growth in SC also increased [PSI +] loss in the ssb1/2∆ strain, this effect was about 170 times less efficient than in the wild-type strain. Thus, prion destabilization in SC medium is due in a significant part to the presence of Ssb and, possibly, due to its release from the ribosome. This indicates that the Ssb-based regulatory circuit is physiologically relevant (Fig. 3b).
Transcription of SSB genes is co-regulated with the ribosomal protein genes, and their expression is decreased in unfavorable conditions when protein synthesis is slowed down (Lopez et al. 1999). Typically, downregulation of overall protein biosynthesis is accompanied by an increased synthesis of specific proteins, making such proteins prone to misfolding and, potentially, to prion formation due to a shortage of Ssb on the ribosomes. It is possible that the release of Ssb to the cytosol in unfavorable conditions represents a general mechanism modulating aggregate formation and protecting cells from potentially toxic self-perpetuating protein aggregates. Such a mechanism would provide an important complement to other regulatory pathways linking stress defense to control of growth and proliferation (for example, see Ho and Gasch 2015).
Notably, RAC components (Zuo1 and Ssz1) are identified among proteins forming insoluble aggregates at heat shock and are solubilized during recovery after return to normal temperature (Wallace et al. 2015). Ssb is also partly shifted to the aggregated fraction in heat-shocked cells, possibly in association with arrested ribosomes; however, a significant fraction of Ssb remains soluble. It is an intriguing possibility that cytosolic Ssb could antagonize propagation of amyloid-like aggregates induced by heat shock, thus preventing accumulation of prions in these conditions. Previous data indicate that heat stress may both induce [PSI +] formation (Tyedmers et al. 2008) and destabilize pre-existing [PSI +] prions (Ali et al. 2014; Chernova et al. 2011; Klaips et al. 2015; Newnam et al. 2011). Further studies should clarify whether or not Ssb release plays a role in these processes.
It is worth mentioning that the RAC defect increases toxicity of a polyQ construct or the construct bearing the QN-rich prion domain (PrD) of the Sup35 protein in the strain bearing a pre-existing endogenous prion which promotes the aggregation of such constructs (Amor et al. 2015). This confirms that the RAC-Ssb complex is indeed involved in detoxification of amyloidogenic misfolded proteins. Moreover, RAC disruption also makes high levels of Sup35 PrD toxic to strains lacking both endogenous prion and the prionogenic protein Rnq1, known to promote prion formation by Sup35 (Amor et al. 2015). This suggests that at high levels of an aggregated protein, uncontrolled non-prion aggregation could be more toxic to the yeast cells than formation of proliferating prions that could be diluted in cell divisions. However, reintroduction of the [PSI +] prion into the strains lacking both Zuo1 and Rnq1 and overexpressing Sup35 PrD restores viability only slightly (Amor et al. 2015). Our data (Kiktev et al. 2015), showing that cytosolic Ssb interferes with prion propagation in zuo1∆ cells, explain why this restoration is so inefficient. Thus, in an artificially generated situation with a highly expressed amyloidogenic protein in a prion-containing culture, the anti-prion action of cytosolic Ssb may become detrimental for the cell.
Potential mechanisms of the antagonism between Ssa and Ssb
While Ssb was not co-purified with the Sup35 prion aggregates in significant amounts, the proportion of Ssa bound to the Sup35 aggregates was decreased by several fold in zuo1∆ or ssz1∆ cells compared to wild-type cells (Kiktev et al. 2015). Thus, the Ssb-RAC disassembly inhibits binding of Ssa to a prion. There could be several (not necessarily mutually exclusive) mechanisms of such inhibition (see Fig. 3b).
-
1.
Direct competition for prion aggregates between Ssa and Ssb. In vitro, both proteins can interact with Sup35 (Allen et al. 2005; Shorter and Lindquist 2008), however, only Ssa is specifically co-purified with Sup35 from extracts of [PSI +] cells (Bagriantsev et al. 2008; Kiktev et al. 2015). It is possible that Ssb outcompetes Ssa for binding to the Sup35 prion polymers, but cannot form stable complexes and thus quickly dissociates from aggregates. While initial binding of a substrate by an Hsp70 protein occurs via SBD, formation of a stable complex with a substrate requires ATPase catalysis by NBD of the Hsp70 (Flynn et al. 1989; Palleros et al. 1991). One possibility is that amyloid-like polymers can stimulate ATP catalysis by Ssa but not by Ssb. Notably, in vitro data show that incorporation of Ssa into Sup35NM aggregates decreases, while incorporation of Ssb increases an anti-aggregation effect of Hsp104 (Shorter and Lindquist 2008). This could be explained by the data showing that “productive” binding of Hsp104 to Sup35 aggregates, resulting in their fragmentation and proliferation, requires Ssa, while “non-productive” binding, resulting in aggregate elimination, does not (Winkler et al. 2012). Possibly, when Ssb outcompetes Ssa for aggregate binding, it cannot promote “productive” binding of Hsp104 and therefore causes a shift of Hsp104 to the “non-productive” pathway.
-
2.
Inhibition or sequestration of Ssa co-chaperones by Ssb. Ssb is known to interact in vivo and/or in vitro with a variety of Ssa cofactors, including cytosolic Hsp40s, Sis1 and Ydj1, nucleotide exchange factors (NEFs) such as Sse1/2 (which also possesses its own chaperone activity), Fes1 and Snl1 (Dragovic et al. 2006; Sharma and Masison 2009; Shorter and Lindquist 2008), although in the cell at least Fes1 preferentially interacts with Ssa (Abrams et al. 2014). Whether or not such a preference is altered when Ssb is released into cytosol remains to be seen. Notably, these Hsp70 cofactors primarily interact with regions other than SBD. For example, nucleotide exchange factors (NEFs) interact with NBD (Shaner et al. 2006; Sharma and Masison 2009). Although the situation with Hsp40 proteins is less clear, at least type II Hsp40 (yeast Sis1) interacts with CTD (Li et al. 2006; Qian et al. 2002). It is possible that cytosolic Ssb binds co-chaperones of Ssa but cannot be activated by them, and therefore is not capable of assisting Hsp104 in prion propagation, thus shifting the balance towards a “non-productive” pathway.
-
3.
Inhibition of Ssa activity by Ssb in a heterodimer. Recent data suggest that DnaK, a bacterial homolog of Hsp70, works as a dimer (Sarbeng et al. 2015). It is possible that yeast Hsp70s also dimerize. If heterodimers between Ssa and Ssb are formed after the release of Ssb into the cytosol, they could be less functional in prion binding, compared to Ssa homodimers.
Although available data are insufficient to distinguish between these explanations, certain clues are provided by the analysis of chaperone/cochaperone interactions and studies of chimeric Ssa-Ssb constructs. Our new data (Kiktev and Chernoff, previously unpublished, Fig. 4a) demonstrate that the amount of Sis1 protein co-purified with His-tagged Ssa is decreased by about two-to threefold in the strains with RAC defects. This agrees with the model proposing that there is competition for co-chaperones between Ssa and cytosolic Ssb, although it is also possible that this could be a secondary effect of decreased binding of Ssa to Sup35 aggregates, as prion-bound Ssa is expected to also be associated with Sis1. Notably, we observed the non-tagged Ssa protein being co-purified with His-tagged Ssa, in agreement with the notion that Ssa can form a dimer (Fig. 4a).

Investigation of possible mechanisms of the Ssa-Ssb antagonism. a Amount of Sis1 pulled down with His-tagged Ssa1 from extracts of RAC-deficient cells is reduced, as compared to the wild-type (WT) cells. The amount of Sis1 in total lysates was the same for all three strains (not shown). Note that non-tagged Ssa (lower band) is co-isolated with His-tagged Ssa1 (upper band), suggestive of the existence of Ssa dimers. b, c Effects of chimeric Ssa/Ssb constructs on [PSI +] propagation in the WT and RAC-deficient strains bearing the “weak” variant of [PSI +] prion. For each construct, indicated under the X axis, results are shown in the following order from left to right: wild-type (WT) strain; zuo1∆ strain; ssz1∆ strain. d, e Effects of chimeric Ssa/Ssb constructs on [PSI +] propagation in the WT and ssb1/2∆ strains bearing the “weak” variant of [PSI +] prion. (Data for WT are repeated from panels b and c respectively). For each construct, indicated under the X axis, results are shown in the following order, from left to right: WT; ssb1/2∆. Chimeric constructs with swapped NBD (first position), SBD (second position) or CTD (third position) of Ssa1 (designated as A) or Ssb1 (designated as B) were expressed from SSA1 (constructs with Ssa NBD) or SSB1 (other constructs) promoters (panels b and d), or from strong P TEF1 promoter (panels c and e). Individual transformants were streaked out, and frequencies of [psi −] colonies were determined. 3–6 independent cultures were tested for each strain/plasmid combination. Bars correspond to standardized errors. The * symbol indicates that the frequency of [psi −] colonies was below 1 %
We have previously shown that SBD is a primary determinant of differences between the effects of Ssa and Ssb on [PSI +] in strains with functional RAC (Allen et al. 2005). We have now extended these studies to strains deficient in RAC complex, in which most Ssb is relocated into the cytosol (Kiktev and Chernoff, previously unpublished data; Fig. 4b, c). Such an approach is more sensitive and enables us to identify some effects that may have been overlooked previously. All chimeric constructs possessing the SBD domain of Ssb1 indeed exhibited a [PSI +] curing effect at levels comparable to complete Ssb1 (or higher) in such a system, with partial exception of BBA bearing NBD and SBD of Ssb1 in combination with CTD of Ssa1. This construct cured [PSI +] efficiently when it was expressed from the strong promoter of the TEF1 gene, but not when it was expressed from the endogenous SSB1 promoter. One possible explanation could be that the replacement of the Ssb CTD region with the respective region from Ssa could partly restore activation of the ATPase activity by a cytosolic co-chaperone (e.g. Sis1).
Notably, we have found out that chimeric constructs BAA and BAB, including the SBD domain of Ssa1, also exhibited a [PSI +]-curing effect that was either noticeable only in the RAC deficient strains or drastically more pronounced in these strains, depending on the construct and promoter. This result is consistent with the potential inhibition in a heterodimer, as computational modeling indicates that the Hsp70 monomers could interact with each other through both NBD–NBD and NBD-SBD interactions (Malinverni et al. 2015). Another explanation could be that cross-talk between NBD and SBD, which is important for promoting an ATP hydrolysis and stabilizing binding, is defective in such chimeric constructs.
An additional observation was that the anti-[PSI +] effects of BAA and BAB proteins were significantly increased in the absence of wild-type Ssb in the strain with functional RAC (Fig. 4d, e). In contrast, no such an increase was detected for the extra copy of wild type Ssb1. Among chimeric constructs with Ssb1 SBD, only BBA showed a consistent but not dramatic increase in [PSI +] curing in the strains lacking wild-type Ssb1, while data for ABA and ABB were inconsistent, demonstrating an increase with one promoter but not with the other. This indicates that in cells with functional RAC, wild-type Ssb interferes with the anti-prion activity of some chimeric constructs, especially in the case of constructs lacking SBD of Ssb1. One plausible explanation of these results is that the ribosome-associated wild-type Ssb forms heterodimers with chimeric proteins and decreases their accumulation in the cytosol, which interferes with their ability to antagonize Ssa and counteract prion propagation. The impact of this interaction is most pronounced for chimeras lacking SBD of Ssb1 and therefore incapable of binding the ribosome-associated nascent polypeptides on their own.
While further experiments are needed to completely elucidate the mechanism of Ssb-Ssa antagonism in prion propagation, our data suggest that this mechanism may involve interactions within the chaperone (and co-chaperone) complexes.
Future perspectives
So far, our knowledge of prion modulation by ribosome-associated chaperones is based on experiments with one yeast prion, [PSI +], which itself is a prion form of a ribosome-interacting protein, the translation termination factor Sup35. Most of the other known yeast prions also require the Hsp104-based chaperone machinery for their propagation, however, they differ in their responses to Hsp104 overproduction, as well as to variations in levels and/or activity of the other components of this machinery, e.g. Ssa and cytosolic Hsp40s (for review, see Chernova et al. 2014; Liebman and Chernoff 2012). While it is expected that the dissociation of Ssb from the ribosome would increase formation of various prions, the effects of cytosolic Ssb on their propagation could vary due to differential responses to the consequences of its interference with Ssa. Experiments aimed at the characterization of the effects of RAC and Ssb on various prions are currently underway.
Notably, recent data suggest that some yeast prions might not be detrimental but rather provide a selective advantage to yeast cells in certain conditions. One example is the [MOT3 +] prion, which is induced by ethanol stress and promotes resistance to high ethanol via facilitating the formation of filamentous “multicellular” assemblies (Holmes et al. 2013). Mechanisms of how growth conditions may promote this formation of potentially “adaptive” prions are poorly understood. The RAC-Ssb complex, sensitive to the conditions influencing translational rates and playing a dual role in prion formation and propagation, is a likely candidate for the link between prions and environmental conditions.
While the orthologs of Zuo1 and Ssz1, named MPP11 and HSP70L1 respectively, are present in mammals and are partly interchangeable with RAC protein in yeast (Jaiswal et al. 2011), the Ssb subfamily is specific for fungi (e.g. see Kominek et al. 2013). It appears that the role of Ssb is played by other member(s) of the Hsp70 family. Is the human ribosome-associated chaperone apparatus as instrumental in regulating human amyloids as its yeast counterpart? Future experiments should answer this question.
References
Abrams JL, Verghese J, Gibney PA, Morano KA (2014) Hierarchical functional specificity of cytosolic heat shock protein 70 (Hsp70) nucleotide exchange factors in yeast. J Biol Chem 289:13155–13167. doi:10.1074/jbc.M113.530014
Aguilar-Calvo P, Garcia C, Espinosa JC, Andreoletti O, Torres JM (2015) Prion and prion-like diseases in animals. Virus Res 207:82–93. doi:10.1016/j.virusres.2014.11.026
Albanese V, Reissmann S, Frydman J (2010) A ribosome-anchored chaperone network that facilitates eukaryotic ribosome biogenesis. J Cell Biol 189:69–81. doi:10.1083/jcb.201001054
Ali M, Chernova TA, Newnam GP, Yin L, Shanks J, Karpova TS, Lee A, Laur O, Subramanian S, Kim D, McNally JG, Seyfried NT, Chernoff YO, Wilkinson KD (2014) Stress-dependent proteolytic processing of the actin assembly protein Lsb1 modulates a yeast prion. J Biol Chem 289:27625–27639. doi:10.1074/jbc.M114.582429
Allen KD, Wegrzyn RD, Chernova TA, Muller S, Newnam GP, Winslett PA, Wittich KB, Wilkinson KD, Chernoff YO (2005) Hsp70 chaperones as modulators of prion life cycle: novel effects of Ssa and Ssb on the Saccharomyces cerevisiae prion [PSI +]. Genetics 169:1227–1242. doi:10.1534/genetics.104.037168
Amor AJ, Castanzo DT, Delany SP, Selechnik DM, van Ooy A, Cameron DM (2015) The ribosome-associated complex antagonizes prion formation in yeast. Prion 9:144–164. doi:10.1080/19336896.2015.1022022
Bagriantsev SN, Gracheva EO, Richmond JE, Liebman SW (2008) Variant-specific [PSI +] infection is transmitted by Sup35 polymers within [PSI +] aggregates with heterogeneous protein composition. Mol Biol Cell 19:2433–2443. doi:10.1091/mbc.E08-01-0078
Blanco LP, Evans ML, Smith DR, Badtke MP, Chapman MR (2012) Diversity, biogenesis and function of microbial amyloids. Trends Microbiol 20:66–73. doi:10.1016/j.tim.2011.11.005
Buxbaum JN, Linke RP (2012) A molecular history of the amyloidoses. J Mol Biol 421:142–159. doi:10.1016/j.jmb.2012.01.024
Chacinska A, Szczesniak B, Kochneva-Pervukhova NV, Kushnirov VV, Ter-Avanesyan MD, Boguta M (2001) Ssb1 chaperone is a [PSI +] prion-curing factor. Curr Genet 39:62–67
Chernoff YO, Newnam GP, Kumar J, Allen K, Zink AD (1999) Evidence for a protein mutator in yeast: role of the Hsp70-related chaperone ssb in formation, stability, and toxicity of the [PSI] prion. Mol Cell Biol 19:8103–8112
Chernova TA et al (2011) Prion induction by the short-lived, stress-induced protein Lsb2 is regulated by ubiquitination and association with the actin cytoskeleton. Mol Cell 43:242–252. doi:10.1016/j.molcel.2011.07.001
Chernova TA, Wilkinson KD, Chernoff YO (2014) Physiological and environmental control of yeast prions. FEMS Microbiol Rev 38:326–344. doi:10.1111/1574-6976.12053
Chiabudini M, Conz C, Reckmann F, Rospert S (2012) Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol Cell Biol 32:4769–4779. doi:10.1128/MCB.00809-12
Chiabudini M, Tais A, Zhang Y, Hayashi S, Wolfle T, Fitzke E, Rospert S (2014) Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol Cell Biol 34:4062–4076. doi:10.1128/MCB.00799-14
Derkatch IL, Bradley ME, Zhou P, Chernoff YO, Liebman SW (1997) Genetic and environmental factors affecting the de novo appearance of the [PSI +] prion in Saccharomyces cerevisiae. Genetics 147:507–519
Derkatch IL, Bradley ME, Hong JY, Liebman SW (2001) Prions affect the appearance of other prions: the story of [PIN +]. Cell 106:171–182
Dragovic Z, Shomura Y, Tzvetkov N, Hartl FU, Bracher A (2006) Fes1p acts as a nucleotide exchange factor for the ribosome-associated molecular chaperone Ssb1p. Biol Chem 387:1593–1600. doi:10.1515/BC.2006.198
Fiaux J, Horst J, Scior A, Preissler S, Koplin A, Bukau B, Deuerling E (2010) Structural analysis of the ribosome-associated complex (RAC) reveals an unusual Hsp70/Hsp40 interaction. J Biol Chem 285:3227–3234. doi:10.1074/jbc.M109.075804
Flynn GC, Chappell TG, Rothman JE (1989) Peptide binding and release by proteins implicated as catalysts of protein assembly. Science 245:385–390
Fowler DM, Koulov AV, Balch WE, Kelly JW (2007) Functional amyloid—from bacteria to humans. Trends Biochem Sci 32:217–224. doi:10.1016/j.tibs.2007.03.003
Gautschi M et al (2001) RAC, a stable ribosome-associated complex in yeast formed by the DnaK-DnaJ homologs Ssz1p and zuotin. Proc Natl Acad Sci USA 98:3762–3767. doi:10.1073/pnas.071057198
Ho YH, Gasch AP (2015) Exploiting the yeast stress-activated signaling network to inform on stress biology and disease signaling. Curr Genet 61:503–511. doi:10.1007/s00294-015-0491-0
Holmes DL, Lancaster AK, Lindquist S, Halfmann R (2013) Heritable remodeling of yeast multicellularity by an environmentally responsive prion. Cell 153:153–165. doi:10.1016/j.cell.2013.02.026
Huang P, Gautschi M, Walter W, Rospert S, Craig EA (2005) The Hsp70 Ssz1 modulates the function of the ribosome-associated J-protein Zuo1. Nat Struct Mol Biol 12:497–504. doi:10.1038/nsmb942
Jaiswal H, Conz C, Otto H, Wolfle T, Fitzke E, Mayer MP, Rospert S (2011) The chaperone network connected to human ribosome-associated complex. Mol Cell Biol 31:1160–1173. doi:10.1128/MCB.00986-10
James P, Pfund C, Craig EA (1997) Functional specificity among Hsp70 molecular chaperones. Science 275:387–389
Kabani M, Melki R (2015a) Sup35p in its soluble and prion states is packaged inside extracellular vesicles. MBio. doi:10.1128/mBio.01017-15
Kabani M, Melki R (2015b) More than just trash bins? Potential roles for extracellular vesicles in the vertical and horizontal transmission of yeast prions. Curr Genet. doi:10.1007/s00294-015-0534-6
Kiktev DA, Melomed MM, Lu CD, Newnam GP, Chernoff YO (2015) Feedback control of prion formation and propagation by the ribosome-associated chaperone complex. Mol Microbiol 96:621–632. doi:10.1111/mmi.12960
Klaips CL, Hochstrasser ML, Langlois CR, Serio TR (2015) Correction: spatial quality control bypasses cell-based limitations on proteostasis to promote prion curing. Elife 4:e06494. doi:10.7554/eLife.06494
Kominek J, Marszalek J, Neuveglise C, Craig EA, Williams BL (2013) The complex evolutionary dynamics of Hsp70s: a genomic and functional perspective. Genome Biol Evol 5:2460–2477. doi:10.1093/gbe/evt192
Koplin A, Preissler S, Ilina Y, Koch M, Scior A, Erhardt M, Deuerling E (2010) A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. J Cell Biol 189:57–68. doi:10.1083/jcb.200910074
Kushnirov VV, Kryndushkin DS, Boguta M, Smirnov VN, Ter-Avanesyan MD (2000) Chaperones that cure yeast artificial [PSI +] and their prion-specific effects. Curr Biol 10:1443–1446
Li J, Wu Y, Qian X, Sha B (2006) Crystal structure of yeast Sis1 peptide-binding fragment and Hsp70 Ssa1 C-terminal complex. Biochem J 398:353–360. doi:10.1042/BJ20060618
Liebman SW, Chernoff YO (2012) Prions in yeast. Genetics 191:1041–1072. doi:10.1534/genetics.111.137760
Lopez N, Halladay J, Walter W, Craig EA (1999) SSB, encoding a ribosome-associated chaperone, is coordinately regulated with ribosomal protein genes. J Bacteriol 181:3136–3143
Malinverni D, Marsili S, Barducci A, De Los Rios P (2015) Large-scale conformational transitions and dimerization are encoded in the amino-acid sequences of Hsp70 haperones. PLoS Comput Biol 11:e1004262. doi:10.1371/journal.pcbi.1004262
Nelson RJ, Ziegelhoffer T, Nicolet C, Werner-Washburne M, Craig EA (1992) The translation machinery and 70 kd heat shock protein cooperate in protein synthesis. Cell 71:97–105
Newnam GP, Wegrzyn RD, Lindquist SL, Chernoff YO (1999) Antagonistic interactions between yeast chaperones Hsp104 and Hsp70 in prion curing. Mol Cell Biol 19:1325–1333
Newnam GP, Birchmore JL, Chernoff YO (2011) Destabilization and recovery of a yeast prion after mild heat shock. J Mol Biol 408:432–448. doi:10.1016/j.jmb.2011.02.034
Nizhnikov AA, Antonets KS, Inge-Vechtomov SG, Derkatch IL (2014) Modulation of efficiency of translation termination in Saccharomyces cerevisiae. Prion 8:247–260. doi:10.4161/pri.29851
Osherovich LZ, Weissman JS (2001) Multiple Gln/Asn-rich prion domains confer susceptibility to induction of the yeast [PSI +] prion. Cell 106:183–194
Palleros DR, Welch WJ, Fink AL (1991) Interaction of hsp70 with unfolded proteins: effects of temperature and nucleotides on the kinetics of binding. Proc Natl Acad Sci USA 88:5719–5723
Protacio RU, Storey AJ, Davidson MK, Wahls WP (2015) Nonsense codon suppression in fission yeast due to mutations of tRNA(Ser. 11) and translation release factor Sup35 (eRF3). Curr Genet 61:165–173. doi:10.1007/s00294-014-0465-7
Prunuske AJ, Waltner JK, Kuhn P, Gu B, Craig EA (2012) Role for the molecular chaperones Zuo1 and Ssz1 in quorum sensing via activation of the transcription factor Pdr1. Proc Natl Acad Sci USA 109:472–477. doi:10.1073/pnas.1119184109
Qian X, Hou W, Zhengang L, Sha B (2002) Direct interactions between molecular chaperones heat-shock protein (Hsp) 70 and Hsp40: yeast Hsp70 Ssa1 binds the extreme C-terminal region of yeast Hsp40 Sis1. Biochem J 361:27–34
Reidy M, Masison DC (2011) Modulation and elimination of yeast prions by protein chaperones and co-chaperones. Prion 5:245–249. doi:10.4161/pri.17749
Rikhvanov EG, Romanova NV, Chernoff YO (2007) Chaperone effects on prion and nonprion aggregates. Prion 1:217–222
Sarbeng EB et al (2015) A functional DnaK dimer is essential for the efficient interaction with Hsp40 heat shock protein. J Biol Chem 290:8849–8862. doi:10.1074/jbc.M114.596288
Shaner L, Sousa R, Morano KA (2006) Characterization of Hsp70 binding and nucleotide exchange by the yeast Hsp110 chaperone Sse1. Biochemistry 45:15075–15084. doi:10.1021/bi061279k
Sharma D, Masison DC (2009) Hsp70 structure, function, regulation and influence on yeast prions. Protein Pept Lett 16:571–581
Shorter J, Lindquist S (2008) Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J 27:2712–2724. doi:10.1038/emboj.2008.194
Tyedmers J, Madariaga ML, Lindquist S (2008) Prion switching in response to environmental stress. PLoS Biol 6:e294. doi:10.1371/journal.pbio.0060294
Wallace EW et al (2015) Reversible, specific, active aggregates of endogenous proteins assemble upon heat stress. Cell 162:1286–1298. doi:10.1016/j.cell.2015.08.041
Werner-Washburne M, Craig EA (1989) Expression of members of the Saccharomyces cerevisiae hsp70 multigene family. Genome 31:684–689
Werner-Washburne M, Stone DE, Craig EA (1987) Complex interactions among members of an essential subfamily of hsp70 genes in Saccharomyces cerevisiae. Mol Cell Biol 7:2568–2577
Wickner RB, Shewmaker FP, Bateman DA, Edskes HK, Gorkovskiy A, Dayani Y, Bezsonov EE (2015) Yeast prions: structure, biology, and prion-handling systems. Microbiol Mol Biol Rev 79:1–17. doi:10.1128/MMBR.00041-14
Willmund F et al (2013) The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 152:196–209. doi:10.1016/j.cell.2012.12.001
Winkler J, Tyedmers J, Bukau B, Mogk A (2012) Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol 198:387–404. doi:10.1083/jcb.201201074
Zhang Y et al (2014) Structural basis for interaction of a cotranslational chaperone with the eukaryotic ribosome. Nat Struct Mol Biol 21:1042–1046. doi:10.1038/nsmb.2908
Acknowledgments
We thank N. Romanova for help in some experiments, D. Cyr for Sis1 antibodies, E. Craig for the gift of chimeric Hsp70 constructs, P. Chandramowlishwaran and R. Howie for the critical reading of the manuscript. This work was supported by Grant MCB 1516872 from National Science Foundation to YOC. Work on chimeric Hsp70 constructs was also supported by St. Petersburg State University via the Grant 14-50-00069 from Russian Science Foundation.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Chernoff, Y.O., Kiktev, D.A. Dual role of ribosome-associated chaperones in prion formation and propagation. Curr Genet 62, 677–685 (2016). https://doi.org/10.1007/s00294-016-0586-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0586-2