
Abstract
We sequenced the entire mitochondrial genome (mitogenome) of two gelechioids, Mesophleps albilinella and Dichomeris ustalella, and compared their genome organization and sequence composition to those of available gelechioid mitogenomes for an enhanced understanding of Gelechioidea genomic characteristics. We compared all available lepidopteran mitogenome arrangements, including that of M. albilinella, which is unique in Gelechioidea, to comprehend the extensiveness and mechanisms of gene rearrangement in Lepidoptera. The genomes of M. albilinella and D. ustalella are 15,274 and 15,410 bp in size, respectively, with the typical sets of mitochondrial (mt) genes. The COI gene begins with CGA (arginine) in all sequenced gelechioids, including M. albilinella and D. ustalella, reinforcing the feature as a synapomorphic trait, at least in the Gelechioidea. Each 353- and 321-bp long A + T-rich region of M. albilinella and D. ustalella contains one (D. ustalella) or two (M. albilinella) tRNA-like structures. The M. albilinella mitogenome has a unique gene arrangement among the Gelechioidea: ARNESF (the underline signifies an inverted gene) at the ND3 and ND5 junction, as opposed to the ARNSEF that is found in ancestral insects. An extensive search of available lepidopteran mitogenomes, including that of M. albilinella, turned up six rearrangements that differ from those of ancestral insects. Most of the rearrangements can be explained by the tandem duplication-random loss model, but inversion, which requires recombination, is also found in two cases, including M. albilinella. Excluding the MIQ rearrangement at the A + T-rich region and ND2 junction, which is found in nearly all Ditrysia, most of the remaining rearrangements found in Lepidoptera appear to be independently derived in that they are automorphic at several taxonomic scales, although current mitogenomic data are limited, particularly for congeneric data.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Insect mitochondrial genomes (mitogenomes) have been widely used in systematics, phylogeography, diagnostics, and molecular evolution (Cameron 2014). Moreover, mitogenome gene rearrangement, independent of gene sequencing, has been used for comparative and evolutionary genomics and phylogenetic inference in a diverse array of taxonomic groups (Boore et al. 1995, 1998; Boore 1999; Curole and Kocher 1999; Rokas and Holland 2000; Dowton et al. 2009; Cameron et al. 2011; Cameron 2014). In particular, the Hymenoptera and hemipteroids, which revealed exceptional and abundant gene rearrangement, have been extensively studied for their diversity, taxonomic extent, and phylogenetic signals of gene rearrangement (Dowton et al. 2002; Cameron 2014).
In contrast, gene rearrangement in Lepidoptera has received little attention. This is because until 2011, all available lepidopteran mitogenome sequences evidenced only one arrangement: the tRNAMet/tRNAIle/tRNAGln (MIQ, underline indicates an inverted gene) at the A + T-rich region and ND2 junction (Kim et al. 2011). This Lepidoptera-specific rearrangement differs from the most common type found in other insects: the IQM arrangement (Boore et al. 1998). However, subsequent investigation by Cao et al. (2012) evidenced the presence of different arrangements in the Lepidoptera. Two species of Hepialoidea, which is one of the most ancient lepidopteran lineages, display the most common insect type, presenting IQM instead of the previously known Lepidoptera-specific rearrangement. This finding consequently reduced the extension of the Lepidoptera-specific rearrangement to the Ditrysia, which includes approximately 98 % of all described Lepidoptera (van Nieukerken et al. 2011). Since that time, new arrangements have been reported from individual species of Lepidoptera, although they are not abundant (e.g., Wang et al. 2014). Nevertheless, lepidopteran arrangements have never previously been scrutinized, although more than 270 mitogenome sequences comprising 44 families in 23 superfamilies (as of August 6, 2015) are GenBank-registered (or published).
The Gelechioidea are distributed worldwide, comprising 18,489 species in 1428 genera, and are the second most species-rich group of Lepidoptera (van Nieukerken et al. 2011). Despite the phylogenetic significance of this mega-diverse superfamily for the understanding of the higher phylogeny of Ditrysia (Kaila et al. 2011), prior to this study, only seven mitogenomic sequences, representing 5 of the 19 families, were available, including only a single species of Gelechiidae in the subfamily Pexicopiinae (Park et al. 2014; Timmermans et al. 2014; Zhao et al. 2014). In fact, due in part to the paucity of taxa included, previous mitogenome-based lepidopteran phylogeny suffered in resolving the relationships of some early-derived groups, including the Gelechioidea (Timmermans et al. 2014). Therefore, additional mitogenomic sequences from a diverse group of Gelechioidea are essential for the inference of interfamilial relationships within Gelechioidea and superfamilial relationships within the Lepidoptera in the future.
In the present study, we sequenced the entire mitogenomes of two Gelechiidae: Mesophleps albilinella, which belongs to the Anacampsinae, and Dichomeris ustalella, which belongs to the Dichomeridinae (Park 1990, 1991; Parsons 1995). M. albilinella is found in Korea and China, whereas D. ustalella is distributed extensively in southeastern Siberia, the Caucasus, Transcaucasia, Korea, Japan, China, Denmark, Belgium, France, and Italy (Park 1990, 1991; Parsons 1995; Li and Sattler 2012). The genome organization and sequence composition of the two mitogenome sequences were compared to those of available gelechioid mitogenomes. We also report that M. albilinella has a unique gene arrangement never previously found in Gelechioidea. The mechanism responsible for this rearrangement appears to involve inversion, which is a rare mechanism (Cameron 2014). Furthermore, the mitogenome sequences of all available lepidopterans were collected from public databases, and their gene arrangements were analyzed to determine the extent of gene rearrangement in Lepidoptera, to infer the major mechanism responsible for genome rearrangements, and to determine the evolutionary independence (or sharing) of any given rearrangement.
Materials and methods
Genomic DNA extraction, PCR, and sequencing
Adult Mesophleps albilinella and Dichomeris ustalella specimens were collected from Geojedo Island, Gyeongsangnam-do Province in Korea on September 25 and August 25, 2012, respectively. Total DNA was extracted from two hind legs using a Wizard™ Genomic DNA Purification Kit according to the manufacturer’s instructions (Promega, USA). The complete mitogenomes were amplified into three overlapping long fragments (LF1–LF3), using genomic DNA as a template, and 26 subsequent overlapping short fragments (SF1–SF 26), using the LFs as templates. The primers for both the LFs and SFs were adapted from Kim et al. (2012), and detailed sequences are presented in Table 1.
LF PCR was performed using LA Taq™ (Takara Biomedical, Japan) under the following conditions: initial denaturation for 2 min at 96 °C, followed by 30 cycles of 10 s at 98 °C and 15 min at 50 °C, and a subsequent 10-min final extension at 72 °C. For SF PCR, AccuPower PreMix (Bioneer, Korea) was used under the following conditions: initial denaturation for 5 min at 94 °C, followed by 35 cycles of 1 min at 94 °C, 1 min at 48–51 °C, and 1 min at 72 °C, with a subsequent final 7-min extension at 72 °C. SF1–SF25 were directly sequenced, whereas SF26, which encompasses the whole A + T-rich region, was sequenced after cloning. Cloning was carried out using a pGEM-T Easy vector (Promega, USA) and HIT-competent cells (Real Biotech Corporation, Taiwan). The resultant plasmid DNA was isolated using a Plasmid Mini Extraction Kit (Bioneer, Korea). DNA sequencing was conducted using an ABI PRISM® BigDye® Terminator v3.1 Cycle Sequencing Kit with an ABI 3100 Genetic Analyzer (PE Applied Biosystems, USA). All products were sequenced from both directions.
Genome annotation
Gene identification, boundary delimitation, and secondary structure predictions for M. albilinella and D. ustalella tRNAs were made using tRNAscan-SE 1.21 with the search mode set as default, Mito/Chloroplast as the search source, invertebrate mitogenomes as the genetic code for tRNA isotype prediction, and a Cove score cutoff of 1 (Lowe and Eddy 1997). By this method, 21 tRNAs were found in both species. tRNASer(AGN), which has a truncated DHU arm, was found in a hand-drawn secondary structure by the alignment of predicted regions of other lepidopteran mitochondrial (mt) tRNASer(AGN), with particular consideration given to the anticodons. MAFFT ver. 6 (Katoh et al. 2002) was used for this process, with the gap opening penalty set to 1.53 and the offset value (≈gap extension penalty) set to 0.5. The individual mt protein-coding gene (PCG) was identified and its boundary delimitated using the blastn program in BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The start and stop codons of the PCGs were further confirmed by the alignment of M. albilinella and D. ustalella PCGs with other lepidopteran mt PCGs, including those of other gelechioids. Two rRNAs and the A + T-rich region were identified and delimitated using the nucleotide blast program in BLAST and further confirmed by alignment with other lepidopteran mt rRNA genes and A + T-rich region sequences. The sequence data were deposited into the GenBank database under the accession numbers KU366707 for M. albilinella and KU366706 for D. ustalella.
Comparative mitochondrial gene analyses
For the genomic comparison, seven gelechioid mitogenomes were downloaded from either GenBank or AMiGA (Feijao et al. 2006) (Table 2). The nucleotide composition of each gene, the whole genome, and the codon positions of the PCGs were calculated using MEGA 6 (Tamura et al. 2013). Translation of nucleotide sequences and calculation of the codon frequency of the PCGs were performed by MEGA 6 based on the invertebrate mt DNA genetic code (Tamura et al. 2013). Gene overlap and intergenic-space sequences were hand-counted. The taxonomic scope of the comparison was limited to the available gelechioids, since the genomic features of this group have never been extensively analyzed.
Results
General perspectives on the M. albilinella and D. ustalella mitogenomes
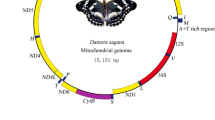
The mitogenomes of M. albilinella and D. ustalella were 15,274 and 15,410 bp in size, respectively, and contained typical sets of mt genes (13 PCGs, 22 tRNAs, and 2 rRNAs) and one major non-coding region, known as the A + T-rich region in insects (Table 2). The extra tRNAs that have been infrequently detected in other Lepidoptera were not found in these species (e.g., Coreana raphaelis and Ctenoptilum vasava in Papilionoidea; Kim et al. 2006; Hao et al. 2012). Twelve PCGs of M. albilinella and D. ustalella started with the typical start codons ATN, but COI began with CGA (arginine) in both M. albilinella and D. ustalella, and all sequenced gelechioids also started with CGA (Fig. 1). The mt PCGs of both M. albilinella and D. ustalella ended with TAA in nine genes, but ended with an incomplete stop codon consisting of a single thymine in four genes (Table 2). Such an incomplete termination codon can become a complete stop codon (TAA) by posttranslational modifications that occur during the mRNA maturation process (Ojala et al. 1981).

Alignment of the initiation context of the COI genes of Gelechioidea, including those of Mesophleps albilinella and Dichomeris ustalella. The amino acid sequences of the first four to six codons are shown on the right-hand side of the figure. Underlined nucleotides indicate the adjacent partial sequence of tRNATyr. Arrows indicate the transcriptional direction. Boxed nucleotides indicate currently proposed translation initiators
Most mt genes of M. albilinella and D. ustalella were well within the size range found in other gelechioids, but a few were slightly larger (Table 2). For example, the size of COIII in M. albilinella was 793 bp, but ranged from 681 to 789 bp in other gelechioids. In addition, the size of tRNAArg in D. ustalella was 70 bp, but ranged from 62 to 68 bp in other gelechioids.
Nucleotide composition
The nucleotide composition of the M. albilinella and D. ustalella mitogenomes was also biased toward A/T nucleotides, at 80.5 and 81.1 %, respectively, similar to other gelechioids, where it ranged from 77.6 % (Oegoconia novimundi) to 81.5 % (Promalactis suzukiella) (Table 3). The A/T content varied profoundly between RNAs (85.4 % in srRNA, 84.1 % in lrRNA, 82.3 % in tRNAs in M. albilinella; and 86.5 % in srRNA, 86.2 % in lrRNA, and 82.2 % in tRNAs in D. ustalella) and PCGs (79.0 % in M. albilinella and 79.3 % in D. ustalella), and this trend was always observed in the sequenced gelechioids (Table 3). The biased usage of A/T nucleotides was also reflected in the form of codon usage (Table 4). Among the 64 available codons, M. albilinella and D. ustalella utilized TTA (Leucine), ATT (Isoleucine), TTT (Phenylalanine), and ATA (Methionine) most frequently, accounting for 40.70 and 40.46 %, respectively. On the other hand, Oegoconia novimundi, belonging to the Autostichidae, had the lowest frequency of the four codons at 35.99 % (Table 4), and this species had the lowest A/T content in the whole genome (77.6 %) as well (Table 3). Currently, O. novimundi is the only species available from this family. Thus, it would be interesting to have more data on this family.
The nucleotide composition of 13 concatenated PCGs in the M. albilinella mitogenome was as follows: A, 33.0 %; T, 46.0 %; C, 10.3 %; and G, 10.7 %. In the D. ustalella mitogenome the composition was A, 33.4 %; T, 45.9 %; C, 10.1 %; and G, 10.7 % (Table 5). The base composition at each codon position of the PCGs in M. albilinella and D. ustalella showed that the third codon position (93.2 and 94.5 %, respectively) harbored a higher A/T content than the first (73.6 and 73.1 %) and second (70.2 and 70.1 %) codon positions, revealing slightly higher content in the first codon position. A similar pattern was also detected in other sequenced gelechioids, with an average of 73.04 % in the first position, 69.88 % in the second position, and 92.69 % in the third position (Table 5).
Non-coding spacer sequences
The M. albilinella and D. ustalella mt genes are interleaved with a total of 151 and 180 bp intergenic spacer sequences, spread over 18 and 16 regions ranging in size from 1 to 54 bp and 1 to 51 bp, respectively (Sup. Table 1). Two relatively longer spacer sequences are noteworthy enough to mention here. One is found between tRNAGln and ND2 at 54 bp in M. albilinella and at 51 bp in D. ustalella. Another is found between tRNASer(UCN) and ND1 at 16 bp in M. albilinella and at 21 bp in D. ustalella (termed Spacers 1 and 2, respectively). Spacer 1 is consistently found in all other gelechioids, at a size ranging from 28 to 66 bp (Sup. Table 1). Spacer 2 is composed of 81.3 and 90.5 % A/T nucleotides in M. albilinella and D. ustalella, respectively, and also is consistently found in all other gelechioids, at sizes ranging from 17 to 40 bp (Sup. Table 1). With the exception of these two longer spacer sequences, M. albilinella has relatively longer sequences at 26 bp. D. ustalella has three sequences longer than 10 bp, but no peculiar aspects were found except that some of these are composed mainly of TA repeats (data not shown). The two gelechioid species had overlapping sequences ranging from 1 to 8 bp spread over 8 (M. albilinella) and 10 (D. ustalella) locations, for a total of 29 bp (M. albilinella) and 27 bp (D. ustalella), respectively (Sup. Table 1).
The A + T-rich region
The lengths of the A + T-rich regions in M. albilinella and D. ustalella were 353 and 321 bp, respectively (Table 2), and were composed of 94.6 and 94.4 % of A/T nucleotides (data not shown). The 369-bp P. suzukiella was the longest and M. albilinella was the third-longest among the gelechioids. The shortest region was found in Pectinophora gossypiella, at 300 bp (Timmermans et al. 2014).
The A + T-rich regions of all gelechioids commonly possess the motif ATAGA close to a 5′-end of the srRNA, with a varying length of poly-T stretch (16 to 19 bp) (Fig. 2a). This motif and the poly-T stretch have been suggested as the site of replication origin of minority strands of mtDNA in the lepidopteran Bombyx mori (Saito et al. 2005). Along with the motif, the A + T-rich regions of most gelechioids, including M. albilinella and D. ustalella, commonly possess the ATTTA sequence, the function of which is unknown; a variable length of TA-repeat (excluding M. albilinella); and a complete or interrupted poly-A stretch immediately upstream of the tRNAMet (excluding P. suzukiella) (Fig. 2a). Two tRNA-like structures in M. albilinella (tRNAMet-like and tRNAAsn-like structures) and one in D. ustalella (tRNALeu-like structure) were found in the A + T-rich region (Fig. 2b).



Structural elements found in the A + T-rich region of Gelechioidea. a Schematic illustration of the A + T-rich region. The presented nucleotides indicate the conserved sequences, such as the ATAGA motif, poly-T stretch, ATTTA sequence, microsatellite-like TA repeat sequence, and poly-A stretch. Dots between sequences indicate omitted sequences b secondary structure of the tRNA-like sequence found in Gelechioidea, including Mesophleps albilinella and Dichomeris ustalella. Subscript indicates the repeat number. The nucleotide position is indicated at the beginning and end sites of the tRNA-like sequence
RNAs
Two rRNA genes (srRNA and lrRNA) were identified at 792 and 1347 bp in M. albilinella and at 795 and 1441 bp in D. ustalella (Table 3). In other gelechioids, the srRNA ranged from 774 (Atrijuglans hetaohei and O. novimundi) to 783 bp (E. eupostica) and the lrRNA ranged from 1299 (Perimede sp.) to 1441 bp (D. ustalella); thus, the present D. ustalella lrRNA is the largest of any known gelechioid lrRNA (Table 3).
Most (21 of 22) tRNAs were folded into a cloverleaf secondary structure, except for a tRNASer(AGN) that lacked the DHU stem in both M. albilinella and D. ustalella (Sup. Figures 1 and 2), as has been shown in many other metazoans (Garey and Wolstenholme 1989). The length of the 22 tRNAs ranged from 64 bp (tRNAArg and tRNAThr) to 72 bp [tRNALeu(CUN)] in M. albilinella, and from 65 bp [(tRNATyr, tRNAGlu, tRNAThr, tRNAPro, and tRNASer(UCN)] to 71 bp (tRNALys) in D. ustalella. The anticodons for each tRNA isotype were identical in all gelechioids, including M. albilinella and D. ustalella (Table 2).
Rearrangement in M. albilinella
The orientation and gene order of the M. albilinella mitogenome differed from any other gelechioid arrangement, including that of D. ustalella (Fig. 3). At the ND3 and ND5 junction, tRNASer(AGN)/tRNAGlu (SE) in the tRNAAla/tRNAArg/tRNAAsn/tRNASer(AGN)/tRNAGlu/tRNAPhe (ARNSEF; underline indicates an inverted gene) cluster region has been rearranged to ES, both with inversion, resulting in ARNESF.

Schematic illustration of the mitochondrial gene rearrangement in Mesophleps albilinella. Gene sizes are not drawn to scale. Gene names that are not underlined indicate a forward transcriptional direction, whereas underlines indicate a reverse transcriptional direction. tRNAs are denoted by one-letter symbols in accordance with the IUPAC-IUB single-letter amino acid codes. Genes and arrangements that are identical to the Ditrysia in Lepidoptera are omitted
Rearrangement in Lepidoptera
Six different mitogenome rearrangements (excluding one arrangement with duplicated tRNA) differing from the ancestral arrangement have been reported among the Lepidoptera, including that of M. albilinella (Fig. 4). The typical lepidopteran arrangement, which is found in the majority of the Ditrysia, differs from the ancestral one found in a variety of insect orders in the “three tRNA region” at the A + T-rich region and ND2 junction. MIQ is the typical lepidopteran arrangement, whereas IQM is the ancestral insect arrangement. This arrangement also is found in the Hepialoidea and Nepticuloidea, which are ancient, non-ditrysian lepidopteran groups (Cao et al. 2012; Timmermans et al. 2014). Apart from “MIQ”, IMQ rearrangement in the three tRNA region is found uniquely in Euripus nyctelius (Nymphalidae in Papilionoidea; Xuan et al. 2015). Another rearranged region in the Lepidoptera is the “ARNSEF cluster region”, which accounts for four of the six lepidopteran rearrangements, including that of M. albilinella (Fig. 4). The Astrotischeria sp., which belongs to one of the ancient, non-ditrysian lepidopteran groups (Tischeriidae in Tischerioidea) was reported to have an RNSAEF rearrangement in this cluster region (Timmermans et al. 2014). Within the Ditrysia, Erynnis montanus (Hesperiidae in Papilionoidea) was reported to have an SN rearrangement, resulting in an ARSNEF (Wang et al. 2014). The remaining two rearrangements, including that of M. albilinella, involve inversion. The ARESNF rearrangement found in Lacosoma valva (Mimallonidae, Mimallonoidea) has inverted genes, along with translocated ones, as compared to ancestral ARNSEF (Timmermans et al. 2014).

Schematic illustration of the available mitochondrial gene arrangements in Lepidoptera. Gene sizes are not drawn to scale. Gene names that are not underlined indicate a forward transcriptional direction, whereas underlines indicate a reverse transcriptional direction. tRNAs are denoted by one-letter symbols in accordance with the IUPAC-IUB single-letter amino acid codes. Dotted lines above the gene names indicate rearranged genes relative to the ancestral arrangement in insects. *Note that Coreana raphaelis (Lycaenidae in Papilionoidea) and Ctenoptilum vasava (Hesperiidae in Papilionoidea) have duplicated tRNASer(AGN) (S1) instead of gene rearrangement
Discussion
Genomic characteristics
The CGA start codon for COI has been regarded as a synapomorphy in the Lepidoptera (Kim et al. 2009), but several exceptions also exist, presenting typical ATN codons (e.g., Ctenoptilum vasava and Lobocla bifasciatus in Hesperiidae; Hao et al. 2012; Kim et al. 2014). Thus, the start codon for COI may not yet be fixed in the Lepidoptera, or a secondary change may be the source of the ATN start codon that is found infrequently in Lepidoptera (Kim et al. 2014). Nevertheless, the conservancy of the start codon as CGA in Gelechioidea may indicate that this feature is a synapomorphic character, at least in the Gelechioidea, if not in all Lepidoptera. However, additional transcriptional data are required to clarify this issue, although recent expressed sequence tag data from a species of Crambidae in the Pyraloidea showed the start codon for COI as CGA (Margam et al. 2011).
With respect to Spacer 1 found between tRNAGln and ND2 (Sup. Table 1), a previous study indicated that this spacer originated in the course of a gene rearrangement, leading to tRNAMet/tRNAIle/tRNAGln (MIQ, underline indicates an inverted gene) in the ditrysian Lepidoptera from the ancestral IQM (Kim et al. 2014). When the ancestral IQM block duplicated, a partial ND2 may have also been duplicated, resulting in IQMIQM-partial ND2. The subsequent deletion process may have accompanied the deletion of the first copy of IQ, second copy of M, and a portion of duplicated ND2, resulting in the MIQ arrangement plus a leftover portion of ND2, such as the 54 bp in M. albilinella and 51 bp in D. ustalella. If this assumption is plausible, there should be some trace of duplication, such as high sequence homology between the leftover portion of ND2 and functional ND2. In fact, sequence alignment of Spacer 1 in gelechioids including M. albilinella and D. ustalella shows substantially high sequence homology to a portion of neighboring ND2, ranging from 58 % (Ethmia eupostica) to 82 % (Perimede sp.), and this identity is obviously higher than can be attributed to chance (Fig. 5). Furthermore, species of Bombycidae, Papilionidae, Pieridae, Lycaenidae, Hesperiidae, Nymphalidae, and Saturniidae also have Spacer1 with substantially high sequence homology to a portion of neighboring ND2 (Kim et al. 2010, 2012, 2014). Spacer 2, located between tRNASer(UCN) and ND1, is known to have a conserved motif sequence, TTAGTAT (Fig. 6), and this sequence has been suggested as the possible recognition site for the transcription termination peptide mtTERM, since it is located just past the final PCG (CytB gene) in the major strand of the mitogenome (Taanman 1999; Cameron and Whiting 2008) and found consistently in all other gelechioids (Fig. 6).

Alignment of the intergenic spacer sequence (Spacer 1) located between tRNAGln and ND2 and neighboring partial ND2 of Gelechioidea. Asterisks indicate consensus sequences in the alignment. Bars (–) were introduced to maximize sequence alignment. Sequence homology between the spacer and the ND2 is shown in the parentheses next to the species name. The nucleotide position is indicated at the beginning and end sites of the sequence

Alignment of the internal spacer region (Spacer 2) located between ND1 and tRNASer(UCN) of the Gelechioidea. The gray-shaded nucleotides indicate the conserved heptanucleotide region (TTAGTAT). Underlined nucleotides indicate the adjacent partial sequences of ND1 and tRNASer(UCN), respectively. Arrows indicate the transcriptional direction
tRNA-like structures in the A + T-rich region have frequently been reported in Lepidoptera (Kim et al. 2009, 2011, 2014; Wan et al. 2013), and we found two tRNA-like structures in M. albilinella and one in D. ustalella (Fig. 2b). These sequences are folded into cloverleaf structures that harbor the proper anticodons and well-matched stem regions, but either the anticodon loop or variable loop contain extra nucleotides. This results in a substantial difference in length and sequence divergence between the regular tRNAs and the tRNA-like pseudo-genes for a given isotype. Our careful analysis of all available gelechioids showed that five of the seven species possessed at least one tRNA-like structure (Fig. 2b). This finding emphasizes the prevalence of tRNA-like pseudogenes in the lepidopteran A + T-rich region. These have been suggested to be spurious tRNAs of random secondary structures, owing to the reduced sequence complexity (>90 % A + T) in this non-coding region (Cameron et al. 2007). Alternatively, the tRNA-like pseudogene in the A + T-rich region has been explained in terms of the failure of the tRNA removal that functions as a primer in replication (Brown et al. 1986; Cantatore et al. 1987).
Rearrangement in Lepidoptera, including M. albilinella
The inverted transposition found in the M. albilinella mitogenome (Fig. 3) can be described through a combination of the tandem duplication-random loss (TDRL) model and recombination (Cameron 2014). The translocation of SE probably occurred by duplication of the SE block, resulting in an SESE arrangement. A subsequent random loss of S in the first copy and E in the second copy may have resulted in the change from SE to ES. Up to this point, this was the rearrangement generated by a typical TDRL model (Mortiz et al. 1987), and the most widely accepted mechanism for mitogenome rearrangements in insects (Cameron 2014; Dowton and Austin 1999). TDRL explains most of the observed mitogenomic rearrangements in insects, but this model cannot explain inversions without recombination (Dowton and Campbell 2001). Thus, the local inversion of ES in M. albilinella may have been caused by a recombination (Dowton and Campbell 2001). The sequential processes that occurred may be double-strand breakage of the mitogenome at either of the two tRNAs (either between N and ES or between ES and F); incorporation of a short, inverted segment of the two tRNAs; and re-association of the breakage, resulting in the inverted “ES” in the ARNESF cluster from the original ARNESF at the ND3 and ND5 junction (Fig. 3). However, the precipitating event between TDRL and inversion remains uncertain.
Among the six different mitogenome rearrangements found in Lepidoptera, the MIQ rearrangement found in Ditrysia, the IMQ rearrangement in E. nyctelius (Nymphalidae in Papilionoidea; Xuan et al. 2015), the RNSAEF rearrangement in Astrotischeria sp. (Timmermans et al. 2014), and the RNSAEF rearrangement in E. montanus (Hesperiidae in Papilionoidea; Wang et al. 2014) can all be explained in terms of TDRL (Mortiz et al. 1987). However, the rearrangements of Lacosoma valva (Mimallonidae in Mimallonoidea; Timmermans et al. 2014) and M. albilinella require inversion and translocation to achieve their current order. In L. valva, the rearrangement appears to involve two independent TDRLs, resulting in ESN, and also two independent inversions resulting in the current ESN (one for the E inversion and the other for the N inversion). Consequently, only two of the six available rearrangements in Lepidoptera involve inversion. These results are consistent with a recent summary on genomic rearrangement in insects indicating that short-range rearrangement by the TDRL model is most common and that inversion is found infrequently (Cameron 2014). In fact, inversion is rarely found in insect groups (e.g., Dermaptera, Hymenoptera, Thysanoptera, Hemiptera, and Phthiraptera) (Shao et al. 2001; Shao and Barker 2003; Thao et al. 2004; Dowton et al. 2009; Wan et al. 2012; Cameron 2014).
Gene rearrangement, which is utilized for phylogenetic markers, is the second major use of mitogenomic data in the evolutionary perspective on insects (Dowton et al. 2003). In particular, the taxonomic extent and synapomorphic status of given rearrangements have received considerable attention, particularly in the Hymenoptera and hemipteroids, which show extremely high rates of gene rearrangement (Cameron 2014). In order to understand the taxonomic extent of gene rearrangement in Lepidoptera, all available complete mitogenome sequences registered in GenBank were obtained (274 mitogenomes from 44 families in 23 superfamilies as of August 6, 2015; Sup. Table 2). The MIQ rearrangement seems to be synapomorphic in the Ditrysia (Cameron 2014), whereas the rearrangement to IMQ from MIQ occurring uniquely in E. nyctelius (Nymphalidae in Papilionoidea; Xuan et al. 2015) can be explained in terms of the secondary loss of the synapomorphic gene arrangement in this particular species. With the exception of the MIQ rearrangement, the rearrangements in Lepidoptera seem to be automorphic at several taxonomic scales, although such an inference is premature, since only limited data are currently available for many taxonomic groups. However, the ARSNEF rearrangement found in E. montanus (Pyrginae, Hesperiidae in Papilionoidea; Wang et al. 2014) is automorphic at the subfamily, family, and superfamily levels in that this rearrangement is unique among 4 species of the same subfamily, 14 species of the same family, and 15 species of the same superfamily. Likewise, the IMQ rearrangement in E. nyctelius (Apaturinae, Nymphalidae in Papilionoidea; Xuan et al. 2015) is automorphic at the subfamily, family, and superfamily levels. However, mitogenome sequences for other congenerics of these two species are not currently available. Thus, the status of the genus-level synapomorphy of the rearrangements remained unanswered in the current study. In addition, the sharing of the ARNESF rearrangement, which was found in a single gelechioid species, M. albilinella (Gelechiidae in Gelechioidea), and another superfamilial member, Rhodopsona rubiginosa (Zygaenidae in Zygaenoidea; Tang et al. 2014), indicates the evolutionary independence of the gene rearrangement. Therefore, the current available data suggest that most gene rearrangement in Lepidoptera is evolutionarily independent, excluding the MIQ rearrangement. This result is consistent with that of a previous investigation of mitogenome arrangement in Hymenoptera, which found that the vast majority of mt gene rearrangements are independently derived (Dowton et al. 2009). Nevertheless, more sequence data from diverse species are obviously required for more robust inference.
In summary, the six different mitogenome rearrangements found in Lepidoptera were explained mainly using TDRL, but the gene rearrangements in M. albilinella, and L. valva involve inversion, indicating that gene inversion does occur in Lepidoptera, although it is rare. Except for the MIQ rearrangement, the remaining rearrangement supports the evolutionary independence in Lepidoptera, indicating the limited utility of gene rearrangement as a phylogenetic marker. Nevertheless, future research focused on congenerics could clarify evolutionary independence at the generic level.
References
Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27:1767–1780
Boore JL, Collins TM, Stanton D, Daehler LL, Brown WM (1995) Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature 376:163–165
Boore JL, Lavrov DV, Brown WM (1998) Gene translocation links insects and crustaceans. Nature 392:667–668
Brown GG, Gadaleta G, Pepe G, Saccone C, Sbisà E (1986) Structural conservation and variation in the D loop-containing region of vertebrate mitochondrial DNA. J Mol Biol 192:503–511
Cameron SL (2014) Insect mitochondrial genomics: implications for evolution and phylogeny. Annu Rev Entomol 59:95–117
Cameron SL, Whiting MF (2008) The complete mitochondrial genome of the tobacco hornworm, Manduca sexta, (Insecta: Lepidoptera: Sphingidae), and an examination of mitochondrial gene variability within butterflies and moths. Gene 408:112–123
Cameron SL, Lambkin CL, Barker SC, Whiting MF (2007) A mitochondrial genome phylogeny of Diptera: whole genome sequence data accurately resolve relationships over broad timescales with high precision. Syst Entomol 32:40–59
Cameron SL, Yoshizawa K, Mizukoshi A, Whiting MF, Johnson KP (2011) Mitochondrial genome deletions and minicircles are common in lice (Insecta: Phthiraptera). BMC Genom 12:394
Cantatore P, Gadaleta MN, Roberti M, Saccone C, Wilson AC (1987) Duplication and remodeling of tRNA genes during the evolutionary rearrangement of mitochondrial genomes. Nature 329:853–855
Cao YQ, Ma C, Chen JY, Yang DR (2012) The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: the ancestral gene arrangement in Lepidoptera. BMC Genom 13:276
Curole JP, Kocher TD (1999) Mitogenomics: digging deeper with complete mitochondrial genomes. Trends Ecol Evol 14:394–398
Dowton M, Austin AD (1999) Evolutionary dynamics of a mitochondrial rearrangement ‘hotspot’ in the Hymenoptera. Mol Biol Evol 16:298–309
Dowton M, Campbell NJH (2001) Intramitochondrial recombination: is it why some mitochondrial genes sleep around? Trends Ecol Evol 16:269–271
Dowton M, Castro LR, Austin AD (2002) Mitochondrial gene rearrangements as phylogenetic characters in the invertebrates: the examination of genome ‘morphology’. Invertebr Syst 16:345–356
Dowton M, Castro LR, Campbell SL, Bargon SD, Austin AD (2003) Frequent mitochondrial gene rearrangements at the Hymenoptera nad3-nad5 junction. J Mol Evol 56:517–526
Dowton M, Cameron SL, Dowavic JI, Austin AD, Whiting MF (2009) Characterization of 67 mitochondrial tRNA gene rearrangements in the Hymenoptera suggests that mitochondrial tRNA gene position is selectively neutral. Mol Biol Evol 26:1607–1617
Feijao PC, Neiva LS, Azeredo-Espin AML, Lessinger AC (2006) AMiGA: the arthropodan mitochondrial genomes accessible database. Bioinformatics 22:902–903
Garey JR, Wolstenholme DR (1989) Platyhelminth mitochondrial DNA: evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J Mol Evol 28:374–387
Hao J, Sun Q, Zhao H, Sun X, Gai Y, Yang Q (2012) The complete mitochondrial genome of Ctenoptilum vasava (Lepidoptera: Hesperiidae: Pyrginae) and its phylogenetic implication. Comp Funct Genomics 2012:1–13
Kaila L, Mutanen M, Nyman T (2011) Phylogeny of the mega-diverse Gelechioidea (Lepidoptera): adaptations and determinants of success. Mol Phylogenet Evol 61:801–809
Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30:3059–3066
Kim I, Lee EM, Seol KY, Yun EY, Lee YB, Hwang JS, Jin BR (2006) The mitochondrial genome of the Korean hairstreak, Coreana raphaelis (Lepidoptera: Lycaenidae). Insect Mol Biol 15:217–225
Kim MI, Baek JY, Kim MJ, Jeong HC, Kim KG, Bae CH, Han YS, Jin BR, Kim I (2009) Complete nucleotide sequence and organization of the mitogenome of the redspotted apollo butterfly, Parnassius bremeri (Lepidoptera: Papilionidae) and comparison with other lepidopteran insects. Mol Cells 28:347–363
Kim MJ, Wan X, Kim KG, Hwang JS, Kim I (2010) Complete nucleotide sequence and organization of the mitogenome of endangered Eumenis autonoe (Lepidoptera: Nymphalidae). Afr J Biotechnol 9:735–754
Kim MJ, Kang AR, Jeong HC, Kim KG, Kim I (2011) Reconstructing intraordinal relationships in Lepidoptera using mitochondrial genome data with the description of two newly sequenced lycaenids, Spindasis takanonis and Protantigius superans (Lepidoptera: Lycaenidae). Mol Phylogenet Evol 61:436–445
Kim JS, Park JS, Kim MJ, Kang PD, Kim SG, Jin BR, Han YS, Kim I (2012) Complete nucleotide sequence and organization of the mitochondrial genome of eri-silkworm, Samia cynthia ricini (Lepidoptera: Saturniidae). J Asia Pac Entomol 15:162–173
Kim MJ, Wang AR, Park JS, Kim I (2014) Complete mitochondrial genomes of five skippers (Lepidoptera: Hesperiidae) and phylogenetic reconstruction of Lepidoptera. Gene 549:97–112
Li HH, Sattler K (2012) A taxonomic revision of the genus Mesophleps Hübner, 1825 (Lepidoptera: Gelechiidae). Zootaxa 3373:1–82
Lowe TM, Eddy SR (1997) TRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Margam VM, Coates BS, Hellmich RL, Agunbiade T, Seufferheld MJ, Sun W et al (2011) Mitochondrial genome sequence and expression profiling for the legume pod borer Maruca vitrata (Lepidoptera: Crambidae). PLoS One 6:e16444
Mortiz C, Dowling TE, Brown WM (1987) Evolution of animal mitochondrial DNA: relevance for population biology and systematics. Annu Rev Ecol Syst 18:269–292
Ojala D, Montoya J, Attardi G (1981) tRNA punctuation model of RNA processing in human mitochondria. Nature 290:470–474
Park KT (1990) Three new species of genera Brachyacma Meyrick and Arisloielia Hiibner (Lepidoptera I Gelechiidae). J Appl Entomol 29:136–143
Park KT (1991) Gelechiidae (Lepidoptera) from North Korea with description of two new species. Annls Hist-Nat Mus Natn Hung 83:117–123
Park JS, Kim SS, Kim KY, Kim I (2014) Complete mitochondrial genome of Suzuki’s Promolactis moth Promalactis suzukiella (Lepidoptera: Oecophoridae). Mitochondrial DNA Early Online:1–2
Parsons MS (1995) A review of the scarce and threatened ethmiine, stathmopodine and gelechiid moths of Great Britain (No. 16). Joint Nature Conservation Committee, Peterborough
Rokas A, Holland PWH (2000) Rare genomic changes as a tool for phylogenetics. Trends Ecol Evol 15:454–459
Saito S, Tamuea K, Aotsuka T (2005) Replication origin of mitochondrial DNA in insects. Genetics 171:433–448
Shao R, Barker SC (2003) The highly rearranged mitochondrial genome of the plague thrips, Thrips imaginis (Insecta: Thysanoptera): convergence of two novel gene boundaries and an extraordinary arrangement of rRNA genes. Mol Biol Evol 20:362–370
Shao R, Campbell NJH, Barker SC (2001) Numerous gene rearrangements in the mitochondrial genome of the wallaby louse, Heterodoxus macropus (Phthiraptera). Mol Biol Evol 18:858–865
Taanman JW (1999) The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta 1410:103–123
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Tang M, Tan M, Meng G, Yang S, Su X, Liu S, Song W, Li Y, Wu Q, Zhang A, Zhou X (2014) Multiplex sequencing of pooled mitochondrial genomes-a crucial step toward biodiversity analysis using mito-metagenomics. Nucleic Acids Res 42:e166
Thao ML, Baumann L, Baumann P (2004) Organization of the mitochondrial genomes of whiteflies, aphids and pysillids (Hemiptera: Sternorrhyncha). BMC Evol Biol 4:25
Timmermans MJTN, Lees DC, Simonsen TJ (2014) Towards a mitogenomic phylogeny of Lepidoptera. Mol Phylogenet Evol 79:169–178
van Nieukerken EJ, Kaila L, Kitching IJ, Kristensen NP, Lees DC, Minet J et al (2011) Order Lepidoptera Linnaeus, 1758. In: Zhang ZQ (ed) Animal biodiversity: an outline of higher-level classification and survey of taxonomic richness. Zootaxa 3148:212–221
Wan X, Kim MI, Kim MJ, Kim I (2012) Complete mitochondrial genome of the free-living earwig, Challia fletcheri (Dermaptera: Pygidicranidae) and phylogeny of Polyneoptera. PLoS One 7:e42056
Wan X, Kim MJ, Kim I (2013) Description of new mitochondrial genomes (Spodoptera litura, Noctuoidea and Cnaphalocrocis medinalis, Pyraloidea) and phylogenetic reconstruction of Lepidoptera with the comment on optimization schemes. Mol Biol Rep 40:6333–6349
Wang AR, Jeong HC, Han YS, Kim I (2014) The complete mitochondrial genome of the mountainous duskywing, Erynnis montanus (Lepidoptera: Hesperiidae): a new gene arrangement in Lepidoptera. Mitochondrial DNA 25:93–94
Xuan S, Song F, Cao L, Wang J, Li H, Cao T (2015) The complete mitochondrial genome of the butterfly Euripus nyctelius (Lepidoptera: Nymphalidae). Mitochondrial DNA Early Online:1–2
Zhao J, Sun Y, Xiao L, Tan Y, Dai H, Bai L (2014) Complete mitochondrial genome of the pink bollworm Pectinophora gossypiella (Lepidoptera: Gelechiidae). Mitochondrial DNA Early online:1–2
Acknowledgments
This study was financially supported by Chonnam National University, 2013.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflict of interest. For this type of study formal consent is not required. This article does not contain any studies with human participants performed by any of the authors. Informed consent was obtained from all individual participants included in the study.
Additional information
Communicated by M. Kupiec.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Park, J.S., Kim, M.J., Jeong, S.Y. et al. Complete mitochondrial genomes of two gelechioids, Mesophleps albilinella and Dichomeris ustalella (Lepidoptera: Gelechiidae), with a description of gene rearrangement in Lepidoptera. Curr Genet 62, 809–826 (2016). https://doi.org/10.1007/s00294-016-0585-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0585-3