
Abstract
The complete nucleotide sequence of the Morus mongolica chloroplast (cp) genome was reported and characterized in this study. The cp genome is a circular molecule of 158,459 bp containing a pair of 25,678 bp IR regions, separated by small and large single-copy regions of 19,736 and 87,363 bp, respectively. The number and relative positions of the 114 unique genes (80 PCGs, 30 tRNAs, and 4 rRNA genes) are almost identical to Morus indica cp genome. Further detailed comparative analyses revealed one hypervariable region, which is responsible for 88 % of the total variation, and 64 indel events between two individuals. There are 78 simple sequence repeats (SSRs) in M. mongolica cp genome, in which 58 of them are mononucleotide repeats. Comparative analysis with M. indica cp genome indicated 22 SSRs with length polymorphisms and 1 SSR with nucleotide content polymorphism. The phylogenetic analysis of 60 PCGs from 62 cp genomes provided strong support for the monophyletic, single origin of Fabidae (N2-fixing) clade.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The chloroplast (cp) is the photosynthetic organelle, which is hypothesized to have arisen from ancient endosymbiotic cyanobacteria, which provides essential energy for plants and algae. In angiosperms, most cp genomes are typically circular DNA, containing a pair of inverted repeats (IRs), one large single-copy region (LSC) and one small single-copy region (SSC) (Jansen et al. 2005; Wu et al. 2009). The size of cp genomes in higher plants ranges from 120 kb to more than 160 kb due to the expansion of IR regions and evolutionary contractions (Wang et al. 2008). The number of complete sequenced cp genomes has increased significantly, since the first cp genome was determined from Nicotiana tabacum (Shinozaki et al. 1986). The cp genome is useful in plant systematics research because of its maternal inheritance and highly conserved structures, and recently, plant phylogenetic studies on the conservation of cp genes have been conducted using partial or whole cp genomes (Asheesh and Vinay 2012).
Moraceae is cosmopolitan in distribution and contains many deciduous trees that have multiple uses (Nyree et al. 2005). The positions of Moraceae were once accorded to the subclass Hamamelidae (Order: Urticales) (http://plants.usda.gov/), but now, which has been repositioned to Order Rosales in Fabidae (also known as Rosid I) according to some nuclear genes or cp genomes (Su et al. 2014; Zhang et al. 2011). Fabidae is considered an N2-fixing clade and contains the four orders Rosales, Cucurbitales, Fagales, and Fabales. Many phylogenetic analyses of this clade indicated a single origin for the angiosperms’ predisposition to fix symbiotic nitrogen (Soltis et al. 1995; Werner et al. 2015).
Mulberry (Family: Moraceae) is an economically important food crop for the domesticated silkworm. The mulberry genome had been sequenced, and the phylogenetic position of mulberry based on single-copy mulberry genes and other 12 sequenced plants indicated that Moraceae has the closest relationship with Rosaceae (He et al. 2013). Morus mongolica is commonly known as a wild type of mulberry, which is native to China and North Korea, and it is also a very important breeding material because of its strong resistance to drought and cold, as well as its hard wood (Clement and Weiblen 2009). Here, we report the cp genome sequence of M. mongolica, and present a comparative analysis between M. mongolica and M. indica, the only one complete cp genome available in Moraceae (Ravi et al. 2006). The genome structure, insertions and deletions (indels), repeat sequences, gene order, and phylogenetics were analyzed.
Materials and methods
Sample collection, genome sequencing, and assembly
Based on its morphological characteristics, the M. mongolica species, in its natural habitat of the Tsinling Mountains (106°55′19″E, 34°14′29″N), was confirmed by Professors Bairen Zhang (Ankang Institute of Agricultural Sciences) and Yunwu Peng (Ankang University). The plant was then transplanted and preserved in the mulberry field of the Key Sericultural Laboratory of Shaanxi, Ankang University.
Approximately, 20 g fresh leaves were sampled from a single M. mongolica plant, and the cpDNA was extracted using a modified high salt method (Shi et al. 2012). After the cpDNA isolation, approximately, 5–10 μg of cpDNA was sheared, followed by adapter ligation and library amplification. Then, the fragmented cpDNAs were subjected to Illumina Sample Preparation Instructions, and single-read sequenced using the Illumina Hiseq 2000 platform. The obtained sequences were assembled using SOAP de novo software (Luo et al. 2012) and reference-based approaches in parallel. The obtained cp genome regions with ambiguous alignments were manually trimmed and considered as gaps. Gaps were filled using the read overhangs at margins and the PCR method. The process was repeated fold, and the minimum fold of the final assembled M. mongolica cpDNA reached approximately 1126-fold.
Genome annotation
The tRNA, rRNA, and protein-coding genes (PCGs) in the assembled genome were predicted and annotated using Dual Organellar GenoMe Annotator with default parameters (Wyman et al. 2004). The locations of tRNAs were then confirmed using tRNAscan-SE software, version 1.21, specifying mito/cpDNA as the source (Lowe and Eddy 1997). The rRNA genes were verified using BLASTN searches against the database of published cp genomes. The positions of the start and stop codons, or intron junctions of PCGs, were verified using the BLASTN searches and sequin program with Plastid genetic code. The gene map was drawn using OGDraw v1.2 (Lohse et al. 2007).
Genome comparison and sequence analysis
To identify the differences between the M. mongolica and M. indica cp genome, the pairwise alignments of the two sequences were performed using Clustal X 1.83. MISA was used to detect the simple sequence repeats (SSRs) with minimal repeat numbers of 10, 5, 4, 3, 3, and 3 for mono-, di-, tri-, tetra-, penta-, and hexa-nucleotides, respectively (Thiel et al. 2003).
Phylogenetic analysis
To illustrate the phylogenetic relationship of the Fabidae clade with other major Rosid clades, the other 61 complete cp genomes were downloaded from GenBank (Table S1). Nicotiana tabacum and Solanum bulbocastanum from Solanaceae were used as outgroups. Some genes missing from a species were examined in each plastome using BLAST searches (e-value cutoff = 1e−10). Then, 60 PCGs found in all of the species were extracted from the selected cp genomes. The amino acid sequences of each of the 60 cp PCGs were aligned using MSWAT (http://mswat.ccbb.utexas.edu/) with default settings, and back translated to nucleotide sequences. Phylogenetic analyses were performed using the concatenated nucleotide sequences and RAxML 7.2.6 software by the maximum likelihood (ML) method (Stamatakis 2006). RAxML searches relied on the General Time Reversible model of nucleotide substitution with the gamma rate model (GTRGAMMA). A bootstrap analysis was performed with 1000 replications using non-parametric bootstrapping as implemented in the “fast bootstrap” algorithm. The absent genes were finally mapped on to the ML phylogenetic tree.
Results
The overall structure and general features of the M. mongolica cp genome
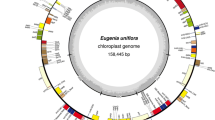
The cp genome of M. mongolica is a closed circular molecule of 158,459 bp (GenBank accession number: KM491711), composed of a pair of IR regions (IRa and IRb) of 25,678 bp, one LSC region of 87,363 bp, and one SSC region of 19,736 bp. It has an overall typical quadripartite structure that resembles the majority of land plant cp genomes. The GC contents of the LSC, SSC, and IR regions, and the whole cp genome are 34.0, 29.3, 42.9, and 36.3 %, respectively (Table 1), which are similar to those of the other reported Rosids cp genomes (Su et al. 2014). The cp genome encodes 133 predicted functional genes, including 88 PCGs of 80,514 bp, and 37 tRNA and 8 rRNA genes of 11,865 bp. Among the 133 genes, 114 are unique genes and 19 are duplicated genes in the IR regions. We found that 16 genes have one intron (10 PCGs and 6 tRNA genes) and 2 PCGs have two introns (clpP and ycf3). Like most other land plants, a maturase K gene (matK) is located within the intron of trnK, rps12 is trans-spliced, with its two 3′ end residues separated by an intron in the IR region, and the 5′ end exon is in the LSC region (Fig. 1). The 8 rRNA genes were composed of two identical copies of 16S-23S-4.5S-5S rRNA gene clusters in the IR region. Each cluster was interrupted by two tRNA genes, trnI and trnA, in the 16S-23S spacer region.

Gene map of the M. mongolica (KM491711), genes lying outside of the outer layer circle are transcribed in the counterclockwise direction, whereas genes inside are transcribed in the clockwise direction. The colored bars indicate known different functional groups. Area dashed darker gray in the inner circle denotes GC content while the lighter gray shows to AT content of the genome. LSC large single-copy, SSC small-single-copy, IR inverted repeat
Comparison of M. mongolica and M. indica cp genomes
A comparative analysis between M. mongolica and M. indica cp genomes revealed that the sequence similarity between the rps4-trnT, trnT-trnL, and trnL-trnF intergenic regions and the intron of trnL is very low (can not be detected using default parameters). Thus, further identification was conducted by sequencing the PCR product of a specifically designed primer pair (Table S2). The results showed that the sequences of M. mongolica and M. indica were 2076 and 1920 bp in length, respectively, which comprised 1.3 and 1.2 % of their genomes (Fig. 1). However, they were responsible for 88 % of the total variation between the two cp genomes. As with the hypervariable region found between Asian and American Equisetum arvense (Kim and Kim 2014), Aegilops speltoides and Aegilops ovata (Guo and Toru 2005), the difference happened mainly in the intergenic spacer (IGS) or intron, and may provide essential information on the evolution of cp phylogenomic studies.
Further, indel analyses in M. mongolica relative to M. indica cp genomes showed that there were 64 indels (217 bp), consisting of 24 insertions (39 bp) and 40 deletions (178 bp). The largest insertion and deletion were 7 and 72 bp, respectively (Table 2). Of the 64 indel events, 41 (64 %) were single base indels, which is similar the percentages in maize and sugarcane (Yamane et al. 2006), and the case in genus citrus, in which, single-base indels are the most frequent ones (Carbonell-Caballero et al. 2015). All of the indel events occurred in the intron and IGS, and had no influence on the gene functions, which showed that non-coding regions were less protected than functional genes.
A total of 293 base substitution events (266 in the LSC region and 27 in the SSC region) and a 2.5:1:0 base substitution numbers per unit length ratio in the LSC:SSC:IR region were found in the M. mongolica cp genome (Table 3), which accounted for 0.18 % of the difference in length. Of the 293 sites, 147 and 146 were transitional (Ts) and transversional changes (Tv), respectively, showing a higher Ts/Tv bias (≈1.0). Meanwhile, the distribution of the base substitutions and the Ts/Tv ratio was uneven. There were 53 in the PCGs with the highest Ts/Tv ratio (2.3), 29 in the introns with a median Ts/Tv ratio (1.6), and 207 in the IGS, which accounted for 71 %, with the lowest Ts/Tv ratio (0.81). In addition, there were four in the tRNA gene. Of the 53 substitutions in the PCGs, 30 were concentrated in psaB (18 Ts and 4 Tv) and rps14 (6 Ts and 2 Tv), and the other 23 substitutions were distributed in 19 genes, with no more than 3 in a gene (Table 3).
Analyses of simple sequence repeats (SSRs)
A total of 78 SSR loci, harboring 742 bp in length, were detected in the M. mongolica cp genome, and there were 58, 6, 2, 10, and 2 mono-, di-, tri-, tetra-, and penta-nucleotide repeats, respectively. Most of the SSRs are mononucleotide repeats, which are consistent with the study of George et al. (2015). All of the mononucleotides and 14 other SSRs were composed of A and T nucleotides, with a higher AT content (99.7 %) in these sequences than in the genome. Among the SSRs, 43 were located in IGS regions and 10 were found in coding genes, including atpF, cemA, ndhF, rpoC2, atpB, rpoB, and ycf1. Compared with M. indica, 52 loci were identity, 22 exhibited length polymorphisms, 3 were not found, and 1 locus exhibited the nucleotide content polymorphism (ATTTC)3 in M. mongolica and (TTTCT)3 in M. indica (Table 4).
Phylogenetic analysis
In this study, the concatenated nucleotide sequences of 60 PCGs of 62 cp genomes of Rosid clade were used to reconstruct the phylogenetic relationships by the ML method. In total, 45,327 nucleotide positions were analyzed, and the best scored ML tree (final ML optimization likelihood = −416381.612810) with bootstrap support values is depicted in Fig. 2. The bootstrap values in most of the nodes (55 of 61) were greater than 90 %, or even 100 % (total 51). Moraceae and Rosaceae were included in Rosales, the sister group of Fagales and Cucurbitales, which is consistent with the early studies (Su et al. 2014; He et al. 2013). The Fabidae clade, which is monophyletic, is composed of the orders Rosales, Cucurbitales, Fagales, and Fabales, and had a single origin for symbiotic nitrogen fixation.

Phylogenetic analysis of the rosid clade using shared PCGs by the ML method, two solanaceae plant is included as the outgroup to root the tree. All bootstrap supports are indicated near the node. The putative events of gene losses are indicated by colored rectangle
Gene content analysis in the selected species showed that six genes had been lost by different degrees. According to Millen et al. (2001), the translation initiation factor 1 infA was lost from the cp during angiosperm evolution. In this study, infA was lost in most of the Rosids, and existed in only Castanopsis echinocarpa and Quercus rubra from Fagales, Cucumis melo subsp. Melo, and Cucumis sativus from Cucurbitales, and Hypseocharis bilobata from Geraniales. The rps16 gene was lost in 15 species, including all the seven from families Chrysobalanaceae and Salicaceae, and one, three, three, and one from Euphorbiaceae, Fabaceae, Brassicaceae, and Geraniaceae, respectively. The rpl22 gene was found to be lost in Fabaceae, Theobroma from Malvaceae, Castanea mollissima, and Trigonobalanus doichangensis from Fagaceae. In addition, rpl32 gene was lost in the three species of Salicaceae, accD was lost in Hypseocharis bilobata from Geraniaceae, and clpP was lost in Viviana marifolia from Vivianiaceae (Fig. 2).
Discussion
The earlier study has shown that M. mongolica is the wild type of mulberry and has a close relationship with M. indica (Yang et al. 2003). In this study, we determined the complete cp genome sequence of M. mongolica, and compared it with the M. indica cp genome. The length of the M. mongolica cp genome was 158,459, 25 bp shorter than that of M. indica, while encoding the identical functional genes in the same order as in M. indica. Further analyses revealed that there was one hypervariable region, 64 indels, 293 base substitutions, accounting for 1.5 % of the total length, between the two cp genomes. The differences between the two cp genomes were distributed mostly in the LSC region, few in the SSC region, and none in IR region, which was consistent with an earlier study’s conclusion that the IR region contributed greatly to the size and conservation of the cp genome (Maier et al. 1995; Guisinger et al. 2010).
The occurrence of base substitutions, also known as SNPs, could be affected by the DNA mismatch repair enzyme, DNA synthetase, and selection pressure (Seo et al. 2000). The ratio of Ts/Tv should be 0.5 in theory, but in fact, the value is often higher than that because of the genetic characteristics of codons, the corresponding pattern of codon replacements, and the genome content (Yang and Yoder 1999; Morton 2003). The ratios in the whole cp genome and IGS region were 1.0 and 0.8, respectively, while the value reached 2.3 in the coding region showing the higher Ts/Tv bias. Further analysis suggests that the AT content is different in the coding and IGS region, 66.9 and 61.0 %, respectively, so nucleotide frequency may be the one possible cause for the Ts/Tv bias (Morton 1995). Also, the cause of bias may be variable among different genes, for example, rps14 (Ts/Tv = 3) has three Ks and five Kn changes, and psaB (Ts/Tv = 4.5) has 20 Ks and two Kn changes. So, natural selection can be possible cause in some genes (Escalante et al. 1998).
Most cp genomes are quite AT rich (above 60 %), have unevenly distributed AT contents, and conserved regions with lower AT contents (Cai et al. 2006). The features of the M. mongolica cp genome are the same, and the AT contents in the whole cp genome, LSC, SSC, and IR regions are 63.7, 67.0, 70.7, and 57.1 %, respectively, with no changes occurring in the IR region of the two mulberry species. Similarly, the regions with a high AT content harbor more variations, such as the hypervariable region with 71 % and the SSRs with 99.7 % AT content. The SSR polymorphisms between M. mongolica and M. indica were all focused on A or T mutations. Meanwhile, as a gene with a higher AT content in many plants, ycf1 (71.4 % in M. mongolica) is one of the most rapid evolutionary cp genes (Barnard-Kubow et al. 2014). For hypervariable regions, the AT contents of these two sequences were higher (71 and 78 % in M. mongolica and M. indica, respectively) than the whole cp genome (63.7 %), which revealed a relationship between AT content and indels as previously described (Kelchner 2000; Yamane et al. 2006). These phenomena showed that there is a positive correlation between the AT content and sequence divergence, and a bias toward A and T changes over G and C changes in plant cp genomes.
As an effective marker for plant phylogenetic studies, the results of our analysis based on 62 cp genomes were congruent with the earlier studies that deemed the Fabidae as monophyletic (Zhang et al. 2011; He et al. 2013; Su et al. 2014). Zhang et al.(2011) used 2 nuclear genes and 10 cp loci to elaborate the phylogenetic relationships among Rosales, and their research of them indicated that Rosales is a monophyletic, and Moraceae is a sister to Rosaceae. The study of Su et al. (2014) indicated that the sequences from the four orders of Fabidae form a clade. Fabidae is considered a N2-fixing clade in many phylogenetic analyses, and the predisposition for symbiotic nitrogen fixation has a single angiosperm origin (Soltis et al. 1995; Werner et al. 2015).
In higher plants, gene loss is an ongoing process. infA, rpl22, rps16, and others have been lost in different degrees in this study. The absence of infA in most species is consistent with the results that the infA gene was found to have been independently transferred to and expressed in the nucleus (Landau et al. 2007; Millen et al. 2001). The rpl22 gene was also lost in Fabaceae and some species of Fagaceae. In the earlier studies of the legume pea, the rpl22 gene was found to be missing from its cp genome, but a functional copy of rpl22 existed in its nucleus. The phylogenetic study indicated that the loss of rpl22 from the cp occurred after it was independently transferred to the nucleus in an ancestor of all flowering plants (Gantt et al. 1991). Studies on the rps16 gene in Medicago truncatula and Populus alba indicate that the loss of rps16 from their cp genome was compensated for by the nuclear-encoded rps16, which can target the cp, as well as mitochondria, and the dual targeting of rps16 to the mitochondria and cp may have emerged before the divergence of monocots and dicots (Ueda et al. 2008). The transfer and substitution events in some genes may be important processes in the eukaryotic cell’s evolution.
References
Asheesh S, Vinay S (2012) Evolutionary analysis of plants using chloroplast. LAP Lambert Academic Publishing, German
Barnard-Kubow KB, Sloan DB, Galloway LF (2014) Correlation between sequence divergence and polymorphism reveals similar evolutionary mechanisms acting across multiple timescales in a rapidly evolving plastid genome. BMC Evol Biol 14(1):1
Cai Z, Penaflor C, Kuehl JV, Leebens-Mack J, Carlson JE, dePamphilis CW, Boore JL, Jansen RK (2006) Complete plastid genome sequences of Drimys, Liriodendron, and Piper: implications for the phylogenetic relationships of magnoliids. BMC Evol Biol 6:77
Carbonell-Caballero J, Alonso R, Ibanez V, Terol J, Talon M, Dopazo J (2015) A phylogenetic analysis of 34 chloroplast genomes elucidates the relationships between wild and domestic species within the genus citrus. MBE. doi:10.1093/molbev/msv082
Clement WL, Weiblen GD (2009) Morphological evolution in the mulberry family (Moraceae). Syst Bot 34(3):530–552
Escalante AA, Lal AA, Ayala FJ (1998) Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics 149(1):189–202
Gantt JS, Baldauf SL, Calie PJ, Weeden NF, Palmer JD (1991) Transfer of rpl22 to the nucleus greatly preceded its loss from the chloroplast and involved the gain of an intron. EMBO 10(10):3073–3078
George B, Bhatt BS, Awasthi M, George B, Singh AK (2015) Comparative analysis of microsatellites in chloroplast genomes of lower and higher plants. Curr Genet. doi:10.1007/s00294-015-0495-9
Guisinger MM, Chumley TW, Kuehl JV, Boore JL, Jansen RK (2010) Implications of the plastid genome sequence of Typha (Typhaceae, Poales) for understanding genome evolution in Poaceae. J Mol Evol 70(2):149–166
Guo CH, Toru T (2005) Sequence analysis of a hotspot region in the chloroplast genome of T. aestivum and Aegilops Species. Sci Agric Sin 38(7):1300–1305
He NJ, Zhang C, Qi XW, Zhao SC, Tao Y, Yang GJ, Lee TH, Wang XY, Cai QL, Li D, Lu MZ, Liao ST, Luo GQ, He RJ, Tan X, Xu YM, Li T, Zhao AC, Jia L, Fu Q, Zeng QW, Gao C, Ma B, Liang JB, Wang XL, Shang JZ, Song PH, Wu HY, Fan L, Wang Q, Shuai Q, Zhua JJ, Wei CJ, Zhu-Salzman K, Jin DC, Wang JP, Liu T, Yu MD, Tang CM, Wang ZJ, Dai FW, Chen JF, Liu Y, Zhao ST, Lin TB, Zhang SG, Wang JY, Wang J, Yang HM, Yang GW, Wang J, Paterson AH, Xia QY, Ji DF, Xiang ZH (2013) Draft genome sequence of the mulberry tree Morus notabilis. Nat Commun 4:2445
Jansen RK, Raubeson LA, Boore JL, dePamphilis CW, Chumley TW, Haberle RC, Wyman SK, Alverson AJ, Peery R, Herman SJ, Fourcade HM, Kuehl JV, McNeal JR, Leebens-Mack J, Cui L (2005) Methods for obtaining and analyzing chloroplast genome sequences. Methods Enzymol 395:348–384
Kelchner SA (2000) The evolution of non-coding chloroplast DNA and its application in plant systematics. Ann Missouri Bot Gard 87(4):482–498
Kim HT, Kim KJ (2014) Chloroplast genome differences between Asian and American Equisetum arvense (Equisetaceae) and the origin of the Hypervariable trnY-trnE intergenic spacer. PLoS One 9(8):e103898
Landau A, Díaz Paleo A, Civitillo R, Jaureguialzo M, Prina AR (2007) Two infA gene mutations independently originated from a mutator genotype in barley. J Hered 98(3):272–276
Lohse M, Drechsel O, Bock R (2007) Organellar genome DRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet 52(5–6):267–274
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25(5):955–964
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J et al (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1(1):18. doi:10.1186/2047-217X-1-18
Maier RM, Neckermann K, Igloi GL, Kössel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251(5):614–628
Millen RS, Olmstead RG, Adams KL, Palmer JD, Lao NT, Heggie L, Kavanagh TA, Hibberd JM, Gray JC, Morden CW, Calie PJ, Jermiin LS, Wolfe KH (2001) Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 13:645–658
Morton BR (1995) Neighboring base composition and transversion/transition bias in a comparison of rice and maizechloroplast noncoding regions. PNAS 92(21):9717–9721
Morton BR (2003) The role of context-dependent mutations in generating compositional and codon usage bias in grass chloroplast DNA. J Mol Evol 56(5):616–629
Nyree JC, Zerega NJ, Clement WL, Datwyler SL, Weiblen GD (2005) Biogeography and divergence times in the mulberry family (Moraceae). Mol Phylogenet Evol 37(2):402–416
Ravi V, Khurana JP, Tyagi AK, Khurana P (2006) The chloroplast genome of mulberry: complete nucleotide sequence, gene organization and comparative analysis. Tree Genetics & Genomes 3:49–59
Seo KY, Jelinsky SA, Loechler EL (2000) Factors that influence the mutagenic patterns of DNA adducts from chemical carcinogens. Mutat Res 463(3):215–246
Shi C, Hu N, Huang H, Gao J, Zhao YJ, Gao LZ (2012) An improved chloroplast DNA extraction procedure for whole plastid genome sequencing. PLoS One 7(2):e31468
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T et al (1986) The complete nucleotide sequence of the tobacco chloroplast genome-its gene organization and expression. EMBO 5(9):2043–2049
Soltis DE, Soltis PS, Morgan DR, Swensen SM, Mullin BC, Dowd JM, Martin PG (1995) Chloroplast gene sequence data suggest a single origin of the predisposition for symbiotic nitrogen fixation in angiosperms. PNAS 92(7):2647–2651
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22(21):2688–2690
Su HJ, Hogenhout SA, Ai-Sadi AM, Kuo CH (2014) Complete chloroplast genome sequence of Omani Lime (Citrus aurantiifolia) and comparative analysis within the rosids. PLoS One 9(11):e113049
Thiel T, Michalek W, Varshney RK, Graner A (2003) Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106(3):411–422
Ueda M, Nishikawa T, Fujimoto M, Takanashi H, Arimura S, Tsutsumi N, Kadowaki K (2008) Substitution of the gene for chloroplast RPS16 was assisted by generation of a dual targeting signal. Mol Biol Evol 25(8):1566–1575
Wang RJ, Cheng CL, Chang CC, Wu CL, Su TM, Chaw SM et al (2008) Dynamics and evolution of the inverted repeat-large single copy junctions in the chloroplast genomes of monocots. BMC Evol Biol 8(1):36
Werner GD, Cornwell WK, Sprent JI, Kattge J, Kiers ET (2015) A single evolutionary innovation drives the deep evolution of symbiotic N2-fixation in angiosperms. Nat Commun 5:4087
Wu C, Lai Y, Lin C, Wang Y, Chaw S (2009) Evolution of reduced and compact chloroplastgenomes (cpDNAs) in gnetophytes: selection toward a lower-cost strategy. Mol Phylogenet Evol 52(1):115–124
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20(17):3252–3255
Yamane K, Yano K, Kawahara T (2006) Pattern and rate of indels evolution inferred from whole chloroplast intergenic regions in sugarcane, maize and rice. DNA Res 13(5):197–204
Yang Z, Yoder AD (1999) Estimation of the transition/transversion rate bias and species sampling. J Mol Evol 48(3):274–283
Yang GW, Feng LC, Jing CJ, Yu MD, Xiang ZH (2003) Analysis of genetic structure variance among Mulberry (Morus L.) populations. Acta Sericologica Sin 29(4):323–329
Zhang SD, Soltis DE, Yang Y, Li DZ, Yi TS (2011) Multi-gene analysis provides a well-supported phylogeny of Rosales. Mol Phylogenet Evol 60(1):21–28
Acknowledgments
This work was supported by the Youth Science and Technology New Star Program of Shaanxi Province, China (No. 2013KJXX-96) and Educational Commission of Shaanxi Province of China (No. 14JS003).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kong, W., Yang, J. The complete chloroplast genome sequence of Morus mongolica and a comparative analysis within the Fabidae clade. Curr Genet 62, 165–172 (2016). https://doi.org/10.1007/s00294-015-0507-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-015-0507-9