
Abstract
In this research, we demonstrated a chondroitin sulfate (CHS), polyvinylpyrrolidone (PVP), and 2-acrylamide-2-methylpropane sulphonic acid (AMPS)-based captopril-loaded mucoadhesive hydrogel that efficiently facilitated the sustained release of drug molecules and is developed by radical polymerization technique. The designed hydrogel network is initially analyzed for the influence of different formulation ingredients as well as their quantities on swelling behavior and mucoadhesion capacity. The physical interaction along with biocompatibility of CHS/PVP-co-poly (AMPS) hydrogels is illustrated by utilizing FT-IR and histopathological analysis. Thermal stability, conversion of crystalline nature ingredient to amorphous nature hydrogel matrix, prolonged and sustained in vitro released behavior at 1.2 pH, porous surface morphology, and enhanced mucoadhesive characteristics are ensured by thermal analysis, XRD, drug in vitro released profile, SEM, and ex-vivo mucoadhesive analysis. Interestingly, the findings suggest that CHS/PVP-co-poly (AMPS)-based hydrogel systems are viable candidates for sustained delivery of captopril by enhancing its adherence with the gastric mucus layer and decreasing its dose frequency.
Graphical abstract

Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Crosslinked polymeric three-dimensional networks that are hydrophilic and capable of absorbing water are known as hydrogels [1]. The composition of hydrogels frequently includes polysaccharides like chondroitin sulfate, hyaluronic acid chitosan, cellulose, and alginate have good water absorption this is mainly owing to hydrophilic groups like amide (-CONH), and quaternary amine (-R4N+), hydroxyl (-OH), carboxylic (-COOH), sulphonic acid (-SO3H), primary amine (-CONH2), and ether groups that are present within the backbone of the polymer are ultimately given hydrogels the capacity to absorb water [2]. Due to variations in such functional groups as well as interactions with molecules of water, each polysaccharide may show variable levels of water retention [3]. In recognition of their outstanding hydrophilic qualities, substantial swelling ratio, biocompatibility, along with widespread availability, hydrogels have been successfully applied to and utilized in a variety of fields, including drug delivery systems, tissue engineering, and wound dressing [4]. Despite these hydrogels' benefits, there are several drawbacks, such as poor mechanical stability and durability or a tiny particular surface area, and these restrict their suitability as systems for drug delivery [5]. Hydrogels made from modified natural polymers as well as in combination with one and more synthetic polymers are commonly utilized as pharmaceutical carriers and have garnered a lot of interest as a result of this issue [6]. In this regard, chondroitin sulfate, chitosan, hyaluronic acid starch, as well as cellulose have undergone chemical modification for the purpose of administering drugs.
Among polymers, chondroitin sulfate (CHS) has been significantly studied for mucoadhesive biomedical applications and pharmaceutical delivery systems. CHS is a soluble mucopolysaccharide obtained from animals comprising D-glucuronic acid covalently bonded to N-acetyl-D-galactosamine. It is considered to be a perfect carrier for drug delivery when crosslinked in the presence of a monomer [7]. Chondroitin sulfate (CHS) exhibits high water solubility, multifunctionality, high biocompatibility, and worthy biodegradability, making it an important polymer for pharmaceutical and biomedical inventions [8]. CHS contains free carboxylic acid groups available for modification, making it a more powerful candidate for hydrogel fabrication due to the considerable quantity of water absorbed.
Different structural modification techniques, such as carboxy-alkylation, alkylation, acylation, quaternization, as well as grafting have been used to enhance both the chemical and physical attributes of CHS. The process of linking various molecules to the CHS backbones and polysaccharides is the grafting approach that is most frequently employed to chemically modify them. The framework of CHS can be grafted with a variety of monomers. 2-acrylamido-2-methylpropanoesulfonic acid, acrylamide, acrylic acid, methyl methacrylate, and peptides are a few examples. Numerous physical and chemical techniques can be utilized to initiate the appropriate grafting reaction [9]. In physical approaches, the reaction begins by utilizing a variety of physical therapies, such as microwave radiation [10], UV radiation [11], and gamma rays [12]. On the contrary, a chemical substance, such as ammonium persulfate, potassium persulfate, is employed to initiate the process in chemical procedures.
The 2-acrylamido-2-methylpropanoesulfonic acid (AMPS) is a monomer that is highly considered by researchers because of its immense stability, non-toxic behavior, and wide availability [13, 14]. AMPS is utilized in blending with various polymers to develop different kinds of delivery systems such as hydrogels [15, 16]. A white crystalline powder of acrylonitrile and isobutylene is produced by Ritter’s reaction initiated by strong acid and aqueous media presence. The drug-loaded AMPS-based formulations dissociate in all pH resulting in pH-independent swelling. Pharmaceutical carriers, skin-sensitive electrodes, drug delivery vehicles, and muscle actuators can be prepared with the aid of AMPS-derived hydrogels [17, 18].
Synthetic copolymer polyvinylpyrrolidone (PVP) bears a negative surface charge, having high water solubility used in the preparation of hydrogel because of its low cytotoxicity and biocompatibility [19]. PVP is extensively utilized in drug delivery or biomedical industries for developing pharmaceutical formulations and wound dressing, because of its biocompatible and non-toxic characteristics [20, 21]. PVP is utilized in the fabrication of mucoadhesive microspheres, ophthalmic drug formulations, pH-controlled medication administration, and multi-polymeric composites.
Captopril is indeed an angiotensin-converting enzyme inhibitor that is widely used to treat emergency hypertension [22]. Captopril belongs to biopharmaceutical class I, having high solubility and high permeability [23]. According to a study using (10 mg) 14 C-captopril orally and intravenously, 24 % (oral) and 38 % (intravenous) of the given dose were eliminated over the course of 24 hours as unaltered captopril molecules in urine. One of captopril's most significant downsides is its brief half-life, which necessitates repeated drug administration which restricts its application in chronically ill individuals and results in low patient compliance. The anticipated maximal pharmacological impact of captopril after oral dosing is 1–2 h, having a bioavailability of about 65%, a half-life of 3 hours, and needing to be administered frequently in 25 to 50 mg doses, 2-3 times per day [24]. On the other hand, it is unstable throughout the intestinal pH, whereas stable at gastric pH (1.2), so that particularly absorbed from the stomach. Captopril absorption is frequently enhanced by carefully deciding where should the dosage form released the drug in the GI tract for a longer period [25]. To get over this problem, sustained release formulation must be developed. Studies show that mucoadhesive sustained release formulations result in sustained inhibition of ACE by captopril [26]. Owing to the sustained and mucoadhesive properties of the polymeric hydrogel, the captopril was kept at the place of absorption for a longer time in GIT. As a result, the dose and frequency of administration are decreased along with boosting the drug's bioavailability [27].
In the present study, CHS/PVP infrastructure was designed and synthesized by combining both natural synthetic polymers with monomers, and intercross-link hydrogel systems were fabricated by using the radical polymerization technique. CHS is the best raw material for developing the mucoadhesive hydrogel matrix, but because the CHS hydrogel structure is delicate, PVP was incorporated to give the formulated matrix more stability. The crosslinker is crucial to the swelling and kinetics of drug release. AMPS offers hydrogels that help with the greatest release of the drug in an acidic media since they are extremely swellable at lower pH levels. IPN hydrogel was given mucoadhesive as well as sustained release properties by mixing CHS, PVP, as well as AMPS. This allowed captopril to endure longer at its point of absorption in the GIT. The dose along with dosing frequency is thereby decreased, while patient compliance is improved. Focusing on the mucoadhesive properties of polymers for sustained release, the impact of CHS on the gelling behavior of PVP blends was investigated. The swelling along with the release behavior of drug molecules from the polymeric system was examined under 1.2 gastric pH. In addition, innovative mucoadhesive sustained polymeric platforms were examined for oral tolerability along with toxicity analysis to illustrate the degree of biocompatibility of developed polymeric hydrogels.
Materials and methods
Materials
Polyvinylpyrrolidone (polymer) (MW = 125,000 g/mol) was bought through Sigma-Aldrich. Chondroitin sulfate (CHS), ammonium peroxodisulfate (APS), sodium bisulfite (SHS), and ethylene glycol dimethacrylate (EGDMA) have been bought from Sigma-Aldrich, UK. 2-acrylamido-2-methylpropanesulfonic acid (AMPS) was bought from Shouguang Pner Chemical Co.; Ltd, China. The captopril (model drug) was received as a research donation from Ferozsons Industries, Pakistan.
Methodologies
CHS/PVP-co-poly (AMPS) hydrogels preparation
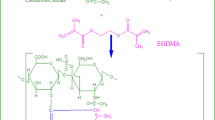
CHS/PVP-co-poly (AMPS) hydrogels were produced via the free-radical polymerization method with some modifications to previously revealed procedures [28,29,30]. After quite a few trial-and-error methods, the concentration utilized for the preparation of hydrogels is indexed in Table 1. A determined polymer (CHS) quantity was solubilized in a certain quantity of distilled water with the aid of using nonstop stirring at 50 °C (optimal temperature), wherein CHS dissolves to form a solution after which the polymer solution is purged with nitrogen for half-hour to eliminate dissolved oxygen. Aqueous solutions of each component were prepared separately including the polymer (PVP), monomer (AMPS), and APS (used as initiator). The PVP solution was slowly poured into the CHS solution and constantly stirred for a given time frame. SHS (another initiator) was incorporated into the APS solution under nonstop stirring. The monomer solution was positioned on a magnetic stirrer; then, the initiator solution was added. The resultant solution of monomer and initiator was combined with the earlier polymer solution, and EGDMA (crosslinker) was added dropwise while stirring continuously. The final volume was made up by the addition of water. Test tubes were filled with the prepared solution, labeled, and transferred to a warm water bath at 45 °C. To evade effervescent and self-acceleration, the temperature was amplified step by step from 45 °C to 60 °C (5 °C per hour) and kept controlled at 60 °C. After 24 h, clear, transparent gels were formed and were withdrawn from the water bath and then, allowed to cool at ambient temperature. CHS/PVP-co-poly (AMPS) hydrogel samples were rinsed with a combination of 1:1 ratio of water and ethanol, positioned in labeled Petri dishes, then dried at approximately 40 °C for 72 h in a hot air oven. The schematic presentation of the CHS/PVP-co-poly (AMPS)-based hydrogels is given in Fig. 1.

Schematic illustration of CHS/PVP-co-poly (AMPS) hydrogels
Drug loading
The diffusion-assisted swelling approach has been selected for the captopril drug loading technique in hydrogel preparation. A pH 1.2 buffer was used to formulate a 1% w/v drug solution. The hydrogel disk of the synthesized hydrogels was weighed and immersed within the solution containing the drug and kept at room temperature for 72 h to attain drug penetration. Subsequently, after 72 h, the disks were swelled and were removed from the drug solution. Deionized water was utilized for washing, and ultimately, the loaded disks were placed in a hot air oven at approximately 40 °C for complete drying [29].
CHS/PVP-co-poly (AMPS) hydrogel characterizations
Fourier transform infrared spectroscopic (FT-IR) analysis
The FTIR spectra of CHS, PVP, AMPS, captopril, as well as drug-loaded hydrogel were predicted. Powdered samples were formed and stored sealed in glass vials for further investigation (Bunkers Germany) within the 4000–600 cm−1 range [31].
Morphological evaluation
SEM Quanta 400 (Cambridge, UK) was utilized to evaluate the produced hydrogel surface morphology. Dried hydrogels were placed on double-sealed tape in order to reduce the optimal sizes and attached to an aluminum stab. They bear ~300 Å thickness with gold lining and by utilizing a gold sputtering module of a vacuum evaporator [31].
Powder X-ray diffractometry (PXRD)
XRD data were recorded at room temperature through the usage of a Bruker D-8 powder diffractometer (Bruker Karlsruhe, Germany). Glass slides were utilized to place the sample in a sample holder. By the usage of radiation from copper, Kα supplied the wavelength of 1.542 Å; then, 1 mm samples had been examined (10°–50°) at a speed of 1° 2θ/min to note peaks of XRD [32].
Thermal evaluation
Thermal analysis (TA) was done with the resource of the use of conducting thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) by using the TA unit of Q5000 series (West Sussex, UK). Developed hydrogel samples were reduced and extruded via 40 number sieves to attain the preferred particle size range. For analysis, the requisite sample (0.5–5 mg) was located in a pan (platinum 100 μL), which was open-linked to microbalance. All samples were investigated beneath nitrogen gas under the use of heating at 20 °C/min [32].
Sol–gel fraction
To evaluate uncross-linked as well as crosslinked parts of fabricated hydrogel, sol–gel analysis was executed by means of the Soxhlet extraction method. The soluble element of hydrogel is sol, while the insoluble element is called gel. A flask with a round bottom (containing water) connected to a condenser was used to place the accurately measured hydrogel disk kept at 85 °C for about 5 h. After extraction of hydrogel, it was placed in a vacuum oven for complete drying. Equations (1) and (2) were employed to calculate sol–gel portions [33, 34]:
where W1 illustrates the pre-extraction weight, and W2 is the constant post-extraction weight of the hydrogel.
Swelling analysis
All developed formulations undergo swelling studies at 37 °C under 1.2 gastric pH. Fabricated hydrogel disks were carefully weighed and kept in the desired pH media and weighed at regular intervals carefully after blotting them with filter paper. The same procedure was repeated so that equilibrium weight was obtained [35]. Take three readings to confirm the attained weight. Equation (3) is used to calculate the dynamic swelling:
where the final weight is presented by Wsand Wd illustrated the primary hydrogel weight at a time “t.”
Drug loading efficiency
Drug loading performance was determined through the extraction method. To aid in swelling as well as drug release, the developed hydrogels were crushed and immersed in 1.2 gastric pH. Lastly, the absorbance was estimated, and the drug loading percentage was evaluated by Eq. (4) [36]:
In vitro dissolution studies
In vitro model drug release assays were accomplished at gastric pH (1.2) by utilizing the USP dissolution type-II apparatus (Curio; DL-0609) (Memmert, Germany) to identify the substantial drug release. A 900 ml of 1.2 pH buffer at 37 ± 0.1 °C was utilized to carry out the dissolution test. The captopril release was assessed by a UV spectrophotometer adjusted at 205 nm wavelength after the predetermined samples (10 ml) were taken at the predetermined intervals. Moreover, the in vitro release of drug molecules from fabricated formulations was compared with the marketed formulation of captopril “acetopril” (Zafa pharmaceutical) [37].
Drug release kinetic mechanism
Important kinetic mechanisms including zero-order, first-order, Higuchi, and Korsmeyer–Peppas models were then applied to the resultant dissolution data of developed hydrogels, which showed different mechanisms of their drug release [38].
Toxicological studies
This research was carried out in strict accordance with the guidelines, and the Organization for Economic Co-operation and Development (OECD) guidelines was followed to evaluate the toxicity of fabricated hydrogel. Healthy rabbits having a weight ranging from 2.0 to 2.5 kg were acquired from the pharmacology research laboratory of the animal facility, Faculty of Pharmacy, and the Islamia University of Bahawalpur for the experiment. Twelve rabbits were taken and separated into two separate groups named Group I and Group II. Temperature (25 °C) was adjusted with 50 to 60 % RH. Animals were allowed to access freely to water. All rabbits were quarantined for one week prior to treatment. To Group I (control), normal saline was administered. Group II (test) received hydrogel of 5 g/kg body weight. Water and food consumption and physical parameters were observed properly on a daily basis. On the fifteenth day, for biochemical blood analysis, the blood sample was taken, and cervical decapitation of one rabbit from each group was done to remove vital organs for histopathological investigation. The study protocol was reviewed and permitted by the PREC (Pharmaceutical Research Ethics Committee) of Islamia University of Bahawalpur, Pakistan [38, 39].
In vitro mucoadhesive study of CHS/PVP-co-poly (AMPS) hydrogel
The mucoadhesive behavior of designed hydrogels was analyzed by the rotary cylinder method using rabbit gastric mucosa with a 1.2 pH buffer. The prepared hydrogel was cut into fine slices. Freshly removed goat gastric mucosa from the slaughterhouse was used. The gastric mucosa was superglued into the cylinders, and the fabricated hydrogels adhered to the mucosa with slight pressing. A 900 ml of gastric simulated 1.2 pH liquid was filled in the basket and prewarmed to 37 °C. The cylinders rotated at 100 rpm by keeping a constant temperature throughout the study. The time at which the hydrogel films became separated from the gastric mucosa was noted. After 5 min and up to 3 h, respectively, the hydrogels were observed for detachment or disintegration from the gastric mucosal surface [37].
Results and discussion
Synthesis mechanism of CHS/PVP-co-poly (AMPS) hydrogel
The major biopolymer CHS was used to formulate the hydrogel, along with PVP (copolymer), AMPS (monomer), APS/SHS (initiators), and EGDMA (crosslinker). To synthesize a gel with the appropriate capabilities, different amounts of biopolymer, copolymer, monomer, and crosslinker were used. Under the event of heat and an inert environment, it is presumed that SHS reacts with APS to produce bisulfite radicals (HSO3˙ˉ), whereas APS breaks down in the water to produce sulfate radicals (SO4˙ˉ). These anion radicals produce radicals on CHS by removing hydroxyl protons out of it. As a result, it is assumed that CHS radicals were produced via anion radicals within the reaction medium. These CHS radicals react with the AMPS molecules when AMPS is introduced. At this point, the solution began modifying its viscosity, indicating that AMPS had likely been grafted onto the CHS, resulting in polymerization as well as the development of radical CHS-g-poly (AMPS) species. PVP was introduced into the CHS-g-poly (AMPS)-growing polymer chain owing to the formed CHS/PVP-co-poly (AMPS). EGDMA was added in predetermined quantities owing to interlinking the graft copolymers. Since the amounts of APS and SHS are lesser compared to the amounts of EGDMA, it is seen that the quantity of EGDMA is larger in all processes than the estimated produced radicals associated with graft products. The probable reaction is illustrated in Fig. 1. Therefore, it is assumed that when EGDMA was introduced, (i) the graft product's radicals attacked EGDMA at first, and when all of the graft radical reactive sites were linked with the EGDMA (considering inter-crosslinking), then, (ii) the remaining EGDMA molecules reacted with one another and polymerized. Therefore, it was expected that EGDMA will self-polymerize during the reaction in addition to crosslinking the two grafted polymers chain.
The polymerization rate and structure of the synthesized hydrogel were greatly influenced by temperature; therefore, temperature was optimized after trials, and optimum temperature was used for the reaction. At increasing temperatures, the radical polymerization process increased, resulting in enhanced crosslinking density and a more compact, dense, and closely interconnected polymeric hydrogel network [40]. As a result, the hydrogel's structure became less porous as well as its swelling ratio decreased, which reduced the hydrogel's capacity to absorb and retain water [41]. At low temperatures, the initiator's thermal degradation also slowed down, making radical synthesis difficult. In order to develop the hydrogel with the necessary structure and porosity, the ideal temperature was therefore established [42].
FTIR spectroscopy
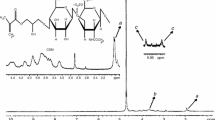
Figure 2 is depicting the FTIR spectrum of captopril, CHS, PVP, AMPS, and the developed drug-loaded hydrogels. The FTIR spectrum of CHS demonstrates a wide band within the range 3602–3201 cm−1 that can be allocated to −OH and NH stretching frequencies. NH stretching modes are superimposed with OH group stretching. The existence of the amide functional group is indicated by a distinctive peak at 1609 cm-1. The bands at 1412 cm-1 and 1377 cm-1 indicate a superposition of C=O bond vibration and the OH vibrations, therefore confirming the presence of a carboxyl group. The vibrational stretching of the sulfate group (S=O) bond was confirmed by the peak at 1227 cm-1, so CHS was confirmed. The stretching mode of CO is indicated with the representative peak at 1028 cm-1. The sharp peak at 2898.83 cm-1 indicating CH stretch showed FTIR spectrum of PVP. A significant spectrum was observed at 1634 cm-1, confirming C=O vibrational stretching. The 1312 cm-1 peak indicates the amide band III. AMPS-FTIR spectra showed structure peaks at 1670.18 cm-1 and 1549.38 cm-1, signifying C=O group stretching (amide I band) and NH bending (amide II band). Characteristic sharp peaks at 1234.17 cm-1 and 1373.78 cm-1 show symmetric, and asymmetric stretching of S=O group confirming AMPS contains the SO3H group. The spectral peak of 2987.91 cm-1 indicated CH stretching of CH2. A strong absorption band around 1078.23 cm-1 to 943.38 cm-1 represents the SOC group [43].

FTIR spectra of PVP, CHS, AMPS, captopril, and loaded (CPA-L) hydrogels
The region of 2977 and 2871 cm-1 has two bands of FTIR spectrum of captopril corresponding to the –CH2 and –CH3 groups. A band at 2563 cm-1 confirmed the occurrence of the -SH group. The vibrational spectra of C=O group and the amide group sharp band were shown in the region of 1749 and 1594 cm-1, respectively [44].
The bond stretching vibrations of the CHS sulfate groups (1227 cm-1) and the AMPS sulfonate group (1373 cm-1) develop band overlapping at 1300 cm-1 of the hydrogel. The captopril loading in the hydrogel is confirmed by a slight deviation of bands and also represents no chemical interaction along with the stability of IPN hydrogel.
Morphological characterization
Figure 3 illustrates the SEM analysis which clarifies that the developed hydrogel has a porous surface, is densely packed and possesses a compact structural behavior, which confirmed the high intermolecular interactions present between the hydrogel contents after the polymerization process. The water diffuses through these pores and, due to this, the hydrogel matrix swelling occurs. So, the high concentration of water penetrated the hydrogel network, followed by the maximum swelling. The rough and porous surface of hydrogel makes them a suitable carrier to load the drug and thus, acts as an efficient drug delivery vehicle [45].

Morphological characteristics of CHS/PVP-co-poly (AMPS) hydrogel
PXRD studies
Figure 4 gives the illustration of the XRD pattern of individual uncross-linked components as well as the developed crosslinked hydrogel. The XRD results of captopril represent prominent peaks at 2θ = 11.21°, 17.29°, 19.05°, 24.98°, and 28.21° [44]. Peaks at 2θ = 39.57° and 45.96° are representative of CHS [46]. Characteristic peaks at 2θ = 10.90°, 12.25°, 14.89°, 20.08°, 21.39°, 22.93°, 24.86°, and 27.43° confirm the AMPS [28]. These peaks' heights can reveal details regarding the existence and arrangement of particular crystalline planes in the structures of polymers, monomers, and drugs. The degree of crystallization of the chemical can be determined by the corresponding intensity of all these peaks. Greater peak intensities imply greater crystallinity. On the contrary, a clear decline in the height of the peaks in the prepared hydrogel is observed, which can be recognized as the formed hydrogel having amorphous nature. The reduction in crystallinity happens due to the formation of chemical bonds among the polymers and the monomer after polymerization The evaluation that was produced as the outcome of the PXRD evaluation agreed with the findings made by [47].

PXRD of captopril, AMPS, CHS, PVP, unloaded (CPA), and drug-loaded (CPA-L) hydrogels
Thermal investigation
Figure 5(a) and (b) shows the pattern obtained from TGA and DSC studies, respectively. Three major stages occur during weight loss, as indicated by the TGA thermogram of CHS. At 102 °C, the first stage begins and sums to 32 % weight reduction until it extends to 207 °C temperature. As the polymeric network is dehydrated and moisture is lost, therefore, the temperature extends. From 207 °C to 258 °C, a second stage exists, which gave weight loss of up to 27 %. The degradation of sulfonate along with carboxylate groups within the polymer backbone causes weight reduction. Whereas, the temperature 258 °C represents the third stage leading to complete polymer degradation [48].

TGA thermogram (a) and DSC curve (b) of CHS, PVP, AMPS, and drug-loaded (CPA-L) hydrogels
Pure PVP gave high stability as 40 % weight loss is seen up to 487 °C. Whereas, two different degradation phases are observed on the thermogram of AMPS. The first phase is a result of water loss at 202.05 °C, while the 273.89 °C temperature is representative of the second phase depending on combustion [33]. Developed hydrogels exhibited initial decay at 200 °C, owing to the evaporation of constrained water. Major degradation starts at 290 °C, exhibiting decay of the functional group (sulfonic acid) as well as side-chain decomposition leaving the primary chain which decays at 440 °C.
After polymerization, the thermal stability of the pristine components has improved, whereas the polymeric network formed is very stable as the components [49]. As a result of crosslinking, the increased thermal stability caused significant intermolecular interaction between the components [50].
According to Fig. 5b, PVP, CHS, AMPS, and designed hydrogels were subjected to DSC analysis. The degradation of the polysaccharide polymer chain gave a prominent endothermic peak at 147 °C and at 334 °C. At 326 °C, exothermic peaks are formed, which represent glass transition temperature (Tg) and oxidative degradation, respectively [46]. The solid-solid transition was observed at 213 °C by PVP due to its non-specific nature during DSC evaluation. Moreover, PVP samples confirm the thermal decomposition by developing endothermic peaks at 103 °C and 395 °C, whereas the sharp exothermic peak was shown at 153 °C and 244 °C [21]. Whereas due to the AMPS melting point, the initial endothermic peak is formed at 196 °C, while 209 °C indicates complete decomposition [38]. DSC graph of CHS/PVP-co-poly (AMPS) exhibits the first endothermic sharp peak at 302 °C, representing the melting of hydrogel, while decomposition was observed at 460 °C.
Sol–gel analysis
Monomer (AMPS), polymers (CHS, PVP), and crosslinkers (EGDMA) were utilized in different ratio, and their impact on the sol–gel fraction was seen (Table 2). In the case of AMPS, the stable gel is formed because the increased monomer and polymer content offer additional free reaction sites for polymerization. In the literature, the gel content was directly proportional to the concentration of polymers and monomers [51, 52]. The sol–gel fraction is further influenced by the amount of crosslinker (EGDMA), as proved by our analysis. More crosslinked structure leads to higher gel content along with higher crosslinker concentration. Thus, as the EGDMA concentration increases, more crosslinking occurs, porosity decreases, and contributes to a physical linkage that developed a tighter network, causing a greater gel fraction [53,54,55].
Swelling studies
To assess the formulation ingredient's impact, the swelling study was performed at 1.2 gastric pH. The swelling pattern was highly affected by the amount of monomer, polymer, and crosslinker as illustrated in Fig. 6. At pH 1.2, the carboxylate groups as well as −SO3− were formed by the ionization of functional groups (amide and carboxylic acid). The expansion in the polymeric network occurs due to decreased hydrogen bonding along with electrostatic repulsion between the amide, carboxylate, and −SO3− groups. Thus, the fabricated hydrogel network tends to swell [56, 57].

Effect of different concentrations of CHS (a), PVP (b), AMPS (c), and EGDMA (d) on swelling index at pH 1.2
At a pH of 1.2, the hydrogel dynamic swelling rises with increasing CHS content from CPA-1 to CPA-3 formulations (Fig. 6a). At low CHS concentrations, monomer molecules readily attach to CHS reactive sites; however, when they entirely possess all accessible reactive sites, steric hindrance produced by polymer chains as well as monomer within reaction inhibits further bonding of monomer with polymer units. [58]. Similarly, another reason for the rise in swelling was the large numbers of carboxylate groups created by CHS, which resulted in a high charge distribution, and strong repulsive interactions were generated between the same charged ions. Thus, the rise in CHS was associated with an increase in swelling [59, 60].
Figure 6b demonstrates an escalation in swelling with a rise in PVP content in CPA-4 to CPA-5, showing the accessibility of more grafting positions at the termination of the polymer. At little PVP concentration, the monomer readily binds to the site of reaction on the polymer. If sites are engaged by monomer, more binding is repressed. This limitation is because steric interruption developed, and the residual monomer becomes unessential in the system. The excess monomer has been converted into a homopolymer and restricted to water absorption. The homopolymer amount lessens as the polymer content rises, giving more active sites for monomer molecules and therefore contributing to swelling [61].
Figure 6c shows the consequence of elevating AMPS on swelling while holding others constant. Swelling increases as AMPS content increases in CPA-6 through CPA-7. As the number of AMPS in the sample rises, the amount of CONH2 and -SO3OH groups upsurge. Water uptake is enhanced as ionization makes groups reactive toward water molecules [62]. Strongly ionizable sulfonate groups of AMPS a hydrophilic monomer fully dissociate over the entire pH range, elevating hydrophilicity and water uptake when AMPS moieties are supplemented with hydrogels [63].
Hydrogels (CPA-9) with increasing content of crosslinking agent (EGDMA) showed little dynamic swelling whenever crosslinking agent content enhanced, the swelling properties of the hydrogel lessened due to the improvement in crosslink density and reduced segment mobility of the exceedingly crosslinked polymer system, demonstrating limited swelling, as displayed in Fig. 6d [64].
Drug loading efficiency
To load the drug in a hydrogel polymeric system, a post-loading method was utilized. The loading efficiency was calculated and tabulated in Table 2. The water absorption property of hydrogel is responsible for drug loading.
At pH 1.2, greater drug loading was observed because of the formation of carboxyl, hydroxyl, and sulfonic acid group ions existing in the CPA polymer chains. Therefore, extra deprotonation of the hydroxyl sulfone and carboxyl functional groups creates further electrostatic repulsion between anionic groups, resulting in increased water absorption. Thus, if there has been increased water uptake into the hydrogels, there will correspondingly be increased drug loading into the hydrogels. In our study, greater swelling and drug loading were observed with increasing polymer (CHS, PVP) and monomer (AMPS) content. However, with increasing crosslinker (EGDMA) content, lower water absorption, as well as drug loading, were observed [65]
Murthy et al. described the phenomenon of swelling and drug entrapment related to the repletion of charged ions. Elevation in deprotonated hydroxyl and carboxyl ions leads to a rise in electrostatic repulsion among the ions [66].
Captopril release profile
The cumulative release of captopril with relation to time in the gastric pH 1.2 is depicted in Fig. 7. The objective was to compare captopril release from fabricated hydrogels and commercially marketed tablets acetopril. The pattern of drug release revealed the sustained drug released from the fabricated hydrogel network. The AMPS pKa = ∼1.9 can explain the phenomena. AMPS dissociates at lower pH, making increasing the system ionicity and rising the electrostatic repulsion between the negatively charged compounds, mainly due to enhanced ionized −SO3− groups, this results in amplified swelling of the hydrogel matrix. The rate of drug release is proportional to the extent of swelling behavior [67].

Drug release profile of all developed hydrogels and commercially available acetopril tablet at pH 1.2
The ratio of monomer, polymer, and crosslinker also affects the release of model drug molecules from the fabricated system. The release rate rises with elevating polymer and monomer concentration as the upsurge in the quantity of ionized −SO3− and −COO− groups. However, increasing the EGDMA content increases the crosslink density, which reduces hydrogel swelling and consequently drug release [68, 69]. With a higher concentration of crosslinker, the matrix showed a limited release of drugs as the network is thick and tightly interconnected. The compact network possesses small pores, which allow limited loading, swelling, and drug release [70, 71]. Drug release from CPA-9 was extended owing to excessive crosslinking and insufficient free radical presence in monomers as well as polymers, which is related to the development of tight linkages and compactness in the polymer system. A high proportion of crosslinking components decreased network expansion along with drug release. In contrast, the in vitro release profile of commercially marketed acetopril tablets indicated 99.74% drug release within 20 min at acidic pH [37].
Kinetic modeling of drug release
The drug release trend of fabricated hydrogel with altering the concentration of CHS, PVP, and AMPS undergoes evaluation through kinetic models, and findings are given in Table 3.
All developed formulations' release profile followed first order as R2 values of the first order were higher at pH 1.2 as compared to zero-order release kinetics, which insured the sustained release of captopril from the formulated CHS/PVP-co-poly (AMPS) hydrogel matrix. Korsmeyer–Peppas outcomes are similar to a regression line with a value of one. Without an expansion process, the model better describes the drug molecules released from the hydrogel. Korsmeyer–Peppas model suggests that the drug release followed an anomalous release mechanism indicating non-Fickian kinetics as the n values varied from 0.507 to 0.922, whereas if the value of n < 0.45, it will follow Fickian kinetics [72]. While regulating water transport, a hydrophilic polymer network turns glassy and potentially releases encapsulated medicines into an aqueous solution. Using a swelling-regulated mechanism of diffusion, a dehydrated hydrogel is capable of simultaneously absorbing water and releasing a hydrophilic drug molecule into the surrounding environment. From the calculated values of n, it was also illustrated that, when the ratio of crosslinker increased the Fickian kinetics was replaced by non-Fickian kinetics. Likewise, by decreasing the monomer content, the value of n was increased and lies in non-Fickian range [72].
Toxicological studies
Various parameters were evaluated in acute oral toxicity studies consisting of general disease symptoms, mortality rate, behavior, water and food intake, salivation, body weight, eye and skin infections. All of these were studied in oral toxicity analysis. Table 4 précises the clinical results of all the aforementioned factors. Clinical evidence has shown that there is no clear alternation in terms of food intake and body weight, and no toxic results have been found.
Since unreacted polymeric components of the crosslinked network can leach out and cause bio-incompatibility therefore, biochemical and hematological findings were adopted to evaluate the toxicity along with biocompatibility of fabricated polymeric matrices with biological systems. Blood serum analysis has been utilized to examine kidney and internal organ functions. The outcomes of the biochemical blood test exhibited slight differences in the results between the groups, but the calculated data were within the normal limits, as shown in Table 5. Comparison of organ weights between test and control groups revealed insignificant notable variation as perceived in our results. Microscopic investigation of the tissue sections showed no evidence of lesion, rupture, degradation, or pathological deviations inside the vital organs, as shown in Fig. 8 [37].

Histopathological tissues examination of different organs from both control and hydrogel-treated groups
In vitro mucoadhesive study of CHS/PVP-co-poly (AMPS) hydrogel
The rotating cylinder approach was utilized to investigate the in vitro mucoadhesion of the fabricated hydrogels with the gastric mucosa. The CHS/PVP-co-poly (AMPS) hydrogel had the most mucoadhesion duration of 130 min, at 0.3 concentration of CHS, as shown in Fig. 9. Because of the low concentration of biopolymer and overhydration, CPA-1 has notably much less mucoadhesion time, i.e., 98 min. The rank order of CHS/PVP-co-poly (AMPS) hydrogel became CPA-3 > CPA-2 > CPA-1. The residue of mucin, sialic acid gets attached to COOH functional group (ionizable single bond), causing the adherence of hydrogel to the mucosal membrane [60, 73]. The adhesive force is enhanced by the uncoiling of the polymer chain under a high number of adhesive components. Therefore, the bioadhesive strength is elevated by enhancing the polymeric concentration (CHS), which causes more interpenetration of polymeric chains into mucin. The mucoadhesive study based on synthetic polymer (Carbapol-934) and natural(CHS)-based fabricated hydrogel utilizing AMPS as monomer via crosslinking polymerization also represents comparable results [53].

In vitro mucoadhesion study of CHS/PVP-co-poly (AMPS) hydrogels
Conclusion
In this research, CHS/PVP-co-poly (AMPS) hydrogels with appropriate mucoadhesion, swelling as well as release profiles were developed by the free radically polymerization approach, by utilizing different feed frame concentrations of CHS, PVP, AMPS, and EGDMA without any observable adverse events after oral administration. The swelling behavior of the produced CHS/PVP-co-poly (AMPS) hydrogel was modifiable by adjusting the polymer and monomer concentration. The fabricated hydrogel demonstrates sustained drug release properties, whereas SEM revealed the porous surface with uniform distribution of drug molecules in the hydrogel matrix. FTIR and XRD revealed that captopril was physically entrapped at the molecular level in the hydrogel network. The designed hydrogel system was shown to be biocompatible and non-toxic during toxicity testing. The mucoadhesive behavior and sustained drug release mechanism proved that the designed IPN hydrogel is more appropriate for a sustained drug delivery system for captopril at the gastric region.
References
Zhang C, Gao K, Chu X, Liu S, Li P, Tang E (2023) Carboxymethyl cellulose/polyamidoamine hydrogel loaded with nano-Ag particles as recoverable efficient catalyst for reduction of 4-nitrophenol in water. Cellulose 30:3729–3743
Manarin E, Corsini F, Trano S, Fagiolari L, Amici J, Francia C, Bodoardo S, Turri S, Bella F, Griffini G (2022) Cardanol-derived epoxy resins as biobased gel polymer electrolytes for potassium-ion conduction. ACS Appl Polym Mater 4:3855–3865
Rahman NA, Hanifah SA, Mobarak NN, Ahmad A, Ludin NA, Bella F, Su’ait MS (2021) Chitosan as a paradigm for biopolymer electrolytes in solid-state dye-sensitised solar cells. Polymer. 230:124092
Arabpour A, Dan S, Hashemipour H (2021) Preparation and optimization of novel graphene oxide and adsorption isotherm study of methylene blue. Arab J Chem 14:103003
Bonomo M, Zarate AS, Fagiolari L, Damin A, Galliano S, Gerbaldi C, Bella F, Barolo C (2023) Unreported resistance in charge transport limits the photoconversion efficiency of aqueous dye-sensitised solar cells: an electrochemical impedance spectroscopy study. Mater Today Sustain 21:100271
Duceac IA, Coseri S (2022) Biopolymers and their derivatives: key components of advanced biomedical technologies. Biotechnol Adv 61:108056
Choudhary A, Sahu S, Vasudeva A, Sheikh NA, Venkataraman S, Handa G, Wadhwa S, Singh U, Gamanagati S, and Yadav S (2021) Comparing effectiveness of combination of collagen peptide type-1, low molecular weight chondroitin sulphate, sodium hyaluronate, and vitamin-C versus oral diclofenac sodium in achilles tendinopathy: a prospective randomized control trial. Cureus 13
Avachat A, Kotwal V (2007) Design and evaluation of matrix-based controlled release tablets of diclofenac sodium and chondroitin sulphate. Aaps Pharmscitech 8:51–56
Agnihotri P, Dan A (2022) Temperature- and pH-responsive hydrogel nanoparticles with embedded au nanoparticles as catalysts for the reduction of dyes. ACS Appl Nano Mater 5:10504–10515
Siccardi S, Amici J, Colombi S, Carvalho JT, Versaci D, Quartarone E, Pereira L, Bella F, Francia C, Bodoardo S (2022) UV-cured self-healing gel polymer electrolyte toward safer room temperature lithium metal batteries. Electrochim Acta 433:141265
Amici J, Torchio C, Versaci D, Dessantis D, Marchisio A, Caldera F, Bella F, Francia C, Bodoardo S (2021) Nanosponge-based composite gel polymer electrolyte for safer Li-O2 batteries. Polymers 13:1625
Elizalde F, Amici J, Trano S, Vozzolo G, Aguirresarobe R, Versaci D, Bodoardo S, Mecerreyes D, Sardon H, Bella F (2022) Self-healable dynamic poly (urea-urethane) gel electrolyte for lithium batteries. J Mater Chem A 10:12588–12596
Ali A, Khalid I, Minhas MU, Barkat K, Khan IU, Syed HK, Umar A (2019) Preparation and in vitro evaluation of Chondroitin sulfate and carbopol based mucoadhesive controlled release polymeric composites of Loxoprofen using factorial design. Eur Polym J. 121:109312
Siddiqua A, Ranjha NM, Rehman S, Shoukat H, Ramzan N, Sultana H (2021) Preparation and characterization of methylene bisacrylamide crosslinked pectin/acrylamide hydrogels. Polym Bull 79:7655–7677
Megías-Vericat JE, Garcia-Robles A, Fernández-Megía MJ, Pérez-Miralles FC, López-Briz E, Casanova B, Poveda JL (2017) Early experience with compassionate use of 2 hydroxypropyl-beta-cyclodextrin for Niemann-Pick type C disease: review of initial published cases. Neurol Sci 38:727–743
Bharate SS (2020) Recent developments in pharmaceutical salts: FDA approvals from 2015 to 2019. Drug Discov Today 26:384–398
Qureshi MA, Khatoon F (2019) Different types of smart nanogel for targeted delivery. J Sci Adv Mater Dev 4:201–212
Atta AM, Dyab AK, Allohedan HA (2013) A novel route to prepare highly surface active nanogel particles based on nonaqueous emulsion polymerization. Polym Adv Technol 24:986–996
Sizílio R, Galvão J, Trindade G, Pina L, Andrade L, Gonsalves J, Lira A, Chaud M, Alves T, Arguelho M (2018) Chitosan/pvp-based mucoadhesive membranes as a promising delivery system of betamethasone-17-valerate for aphthous stomatitis. Carbohydr Polym 190:339–345
Poonguzhali R, Basha SK, Kumari VS (2017) Synthesis and characterization of chitosan-PVP-nanocellulose composites for in-vitro wound dressing application. Int J Biol Macromol 105:111–120
Abdelrazek E, Elashmawi I, Labeeb S (2010) Chitosan filler effects on the experimental characterization, spectroscopic investigation and thermal studies of PVA/PVP blend films. Phys B Condens Matter 405:2021–2027
Sheikh AF, Memon FS, Sahar NU (2022) Angiotensin-converting enzyme inhibitors; their efficacy, adverse effects, and the genetic influence on cough they may cause. Mol Med Commun 2:77–88
Cruz-López EO, Ye D, Wu C, Lu HS, Uijl E, Colafella KMM, Danser AJ (2022) Angiotensinogen suppression: a new tool to treat cardiovascular and renal disease. Hypertension. 79:2115–2126
Iqbal FM, Iqbal S, Nasir B, Hassan W, Ahmed H, Iftikhar SY (2022) Formulation of captopril-loaded hydrogel by microwave-assisted free radical polymerization and its evaluation. Polym Bull 79:7613–7633
Ghumman SA, Noreen S, Hameed H, Elsherif MA, Shabbir R, Rana M, Junaid K, Bukhari SNA (2022) Synthesis of pH-sensitive cross-linked basil seed gum/acrylic acid hydrogels by free radical copolymerization technique for sustained delivery of captopril. Gels 8:291
Tayyab A, Mahmood A, Ijaz H, Sarfraz RM, Zafar N, Danish Z (2022) Formulation and optimization of captopril-loaded microspheres based compressed tablets: in vitro evaluation. Int J Polym Mater Polym Biomater 71:233–245
Sahin B, Ergul M (2022) Captopril exhibits protective effects through anti-inflammatory and anti-apoptotic pathways against hydrogen peroxide-induced oxidative stress in C6 glioma cells. Metab Brain Dis 37:1221–1230
Shoukat H, Pervaiz F, Noreen S, Nawaz M, Qaiser R, Anwar M (2020) Fabrication and evaluation studies of novel polyvinylpyrrolidone and 2-acrylamido-2-methylpropane sulphonic acid-based crosslinked matrices for controlled release of acyclovir. Polym Bull 77:1869–1891
Anwar M, Pervaiz F, Shoukat H, Noreen S, Shabbir K, Majeed A, Ijaz S (2021) Formulation and evaluation of interpenetrating network of xanthan gum and polyvinylpyrrolidone as a hydrophilic matrix for controlled drug delivery system. Polym Bull 78:59–80
Khalid I, Ahmad M, Minhas MU, Barkat K (2018) Synthesis and evaluation of chondroitin sulfate based hydrogels of loxoprofen with adjustable properties as controlled release carriers. Carbohydr Polym 181:1169–1179
Majeed A, Pervaiz F, Shoukat H, Shabbir K, Noreen S, Anwar M (2020) Fabrication and evaluation of pH sensitive chemically cross-linked interpenetrating network [Gelatin/Polyvinylpyrrolidone-co-poly (acrylic acid)] for targeted release of 5-fluorouracil. Polym Bull 79:1–20
Abid U, Pervaiz F, Shoukat H, Rehman S, Abid S (2022) Fabrication and characterization of novel semi-IPN hydrogels based on xanthan gum and polyvinyl pyrrolidone-co-poly (2-acrylamido-2-methyl propane sulfonic acid) for the controlled delivery of venlafaxine. Polym Plast Technol Mater 61:577–592
Shoukat H, Pervaiz F, Rehman S (2022) Pluronic F127-co-poly (2 acrylamido-2-methylpropane sulphonic acid) crosslinked matrices as potential controlled release carrier for an anti-depressant drug: in vitro and in vivo attributes. Chem Pap 76:2917–2933
Qaiser R, Pervaiz F, Shoukat H, Yasin H, Hanan H, Murtaza G (2023) Mucoadhesive chitosan/polyvinylpyrrolidone-co-poly (2-acrylamide-2-methylpropane sulphonic acid) based hydrogels of captopril with adjustable properties as sustained release carrier: formulation design and toxicological evaluation. J Drug Deliv Sci Technol 81:104291
Pervaiz F, Tanveer W, Shoukat H, Rehman S (2022) Formulation and evaluation of polyethylene glycol/Xanthan gum-co-poly (Acrylic acid) interpenetrating network for controlled release of venlafaxine. Polym Bull 80:469–493
Shoukat H, Pervaiz F, Khan M, Rehman S, Akram F, Abid U, Noreen S, Nadeem M, Qaiser R, Ahmad R (2022) Development of β-cyclodextrin/polyvinypyrrolidone-co-poly (2-acrylamide-2-methylpropane sulphonic acid) hybrid nanogels as nano-drug delivery carriers to enhance the solubility of Rosuvastatin: an in vitro and in vivo evaluation. Plos One 17:e0263026
Noreen S, Pervaiz F, Ijaz M, Shoukat H (2022) Synthesis and characterization of pH-sensitive chemically crosslinked block copolymer [Hyaluronic acid/Poloxamer 407-co-poly (Methacrylic acid)] hydrogels for colon targeting. Polym Plast Technol Mater 61:1071–1087
Shoukat H, Pervaiz F, Noreen S, Nawaz M, Qaiser R, Anwar M (2020) Fabrication and evaluation studies of novel polyvinylpyrrolidone and 2-acrylamido-2-methylpropane sulphonic acid-based crosslinked matrices for controlled release of acyclovir. Polym Bull 77:1869–1891
Tabassum M, Pervaiz F, Shoukat H (2022) Fabrication and evaluation of gelatin-PVA-co-poly (2-acrylamido-2-methylpropane sulfonic acid)-based hydrogels for extended-release of sitagliptin and metformin by employing response surface methodology. Chem Pap 76:4081–4097
Godiya CB, Kumar S, Xiao Y (2020) Amine functionalized egg albumin hydrogel with enhanced adsorption potential for diclofenac sodium in water. J Hazardous Mater 393:122417
Banivaheb S, Dan S, Hashemipour H, Kalantari M (2021) Synthesis of modified chitosan TiO2 and SiO2 hydrogel nanocomposites for cadmium removal. J Saudi Chem Soc 25:101283
Dan S, Kalantari M, Kamyabi A, Soltani M (2022) Synthesis of chitosan-g-itaconic acid hydrogel as an antibacterial drug carrier: optimization through RSM-CCD. Polym Bull 79:8575–8598
Rastogi PK, Krishnamoorthi S, Ganesan V (2012) Synthesis, characterization, and ion exchange voltammetry study on 2-acrylamido-2-methylpropane sulphonic acid and N-(hydroxymethyl) acrylamide-based copolymer. J Appl Polym Sci 123:929–935
Stulzer H, Rodrigues P, Cardoso T, Matos J, Silva M (2008) Compatibility studies between captopril and pharmaceutical excipients used in tablets formulations. J Therm Anal Calorim 91:323–328
Barkat K, Ahmad M, Minhas MU, Khalid I, Malik NS (2020) Chondroitin sulfate-based smart hydrogels for targeted delivery of oxaliplatin in colorectal cancer: preparation, characterization and toxicity evaluation. Polym Bull 77:6271–6297
Amrutkar JR, Gattani SG (2009) Chitosan–chondroitin sulfate based matrix tablets for colon specific delivery of indomethacin. AAPS pharmscitech 10:670–677
Guo Y, Shalaev E, Smith S (2013) Physical stability of pharmaceutical formulations: solid-state characterization of amorphous dispersions. Trends Analyt Chem 49:137–144
Wang L-F, Shen S-S, Lu S-C (2003) Synthesis and characterization of chondroitin sulfate–methacrylate hydrogels. Carbohydr Polym 52:389–396
Hu X, Feng L, Wei W, Xie A, Wang S, Zhang J, Dong W (2014) Synthesis and characterization of a novel semi-IPN hydrogel based on Salecan and poly (N, N-dimethylacrylamide-co-2-hydroxyethyl methacrylate). Carbohydr Polym 105:135–144
Ray M, Pal K, Anis A, Banthia A (2010) Development and characterization of chitosan-based polymeric hydrogel membranes. Des Monomers Polym 13:193–206
Yin L, Fei L, Cui F, Tang C, Yin C (2007) Superporous hydrogels containing poly (acrylic acid-co-acrylamide)/O-carboxymethyl chitosan interpenetrating polymer networks. Biomaterials 28:1258–1266
Ullah K, Khan SA, Murtaza G, Sohail M, Manan A, Afzal A (2019) Gelatin-based hydrogels as potential biomaterials for colonic delivery of oxaliplatin. Int J Pharm 556:236–245
Ali L, Ahmad M, Usman M (2015) Evaluation of cross-linked hydroxypropyl methylcellulose graft-methacrylic acid copolymer as extended release oral drug carrier. Cell Chem Technol 49:143–151
Khanum H, Ullah K, Murtaza G, Khan SA (2018) Fabrication and in vitro characterization of HPMC-g-poly (AMPS) hydrogels loaded with loxoprofen sodium. Int J Biol Macromol 120:1624–1631
Ranjha NM, Ayub G, Naseem S, Ansari MT (2010) Preparation and characterization of hybrid pH-sensitive hydrogels of chitosan-co-acrylic acid for controlled release of verapamil. J Mater Sci Mater Med 21:2805–2816
Durmaz S, Okay O (2000) Acrylamide/2-acrylamido-2-methylpropane sulfonic acid sodium salt-based hydrogels: synthesis and characterization. Polymer 41:3693–3704
Sohail M, Ahmad M, Minhas MU, Ali L, Khalid I, Rashid H (2015) Controlled delivery of valsartan by cross-linked polymeric matrices: synthesis, in vitro and in vivo evaluation. Int J Pharm 487:110–119
Lee C-T, Kung P-H, Lee Y-D (2005) Preparation of poly (vinyl alcohol)-chondroitin sulfate hydrogel as matrices in tissue engineering. Carbohydr Polym 61:348–354
Suhail M, Ullah H, Vu QL, Khan A, Tsai M-J, Wu P-C (2022) Preparation of pH-responsive hydrogels based on chondroitin sulfate/alginate for oral drug delivery. Pharmaceutics 14:2110
Suhail M, Wu P-C, Minhas MU (2021) Development and characterization of pH-sensitive chondroitin sulfate-co-poly(acrylic acid) hydrogels for controlled release of diclofenac sodium. J Saudi Chem Soc 25:101212
Finkenstadt VL, Willett JL (2005) Reactive extrusion of starch-polyacrylamide graft copolymers: effects of monomer/starch ratio and moisture content. Macromol Chem Phys 206:1648–1652
Pourjavadi A, Barzegar S, Zeidabadi F (2007) Synthesis and properties of biodegradable hydrogels of κ-carrageenan grafted acrylic acid-co-2-acrylamido-2-methylpropanesulfonic acid as candidates for drug delivery systems. React Funct Polym 67:644–654
Atta AM (2002) Swelling behaviors of polyelectrolyte hydrogels containing sulfonate groups. Polym Adv Technol 13:567–576
Ramelow US, Pingili S (2010) Synthesis of ethylene glycol dimethacrylate-methyl methacrylate copolymers, determination of their reactivity ratios, and a study of dopant and temperature effects on their conductivities. Polymers 2:265–285
Hussain T, Ranjha NM, Shahzad Y (2011) Swelling and controlled release of tramadol hydrochloride from a pH-sensitive hydrogel. Des Monomers Polym 14:233–249
Murthy PK, Mohan YM, Sreeramulu J, Raju KM (2006) Semi-IPNs of starch and poly (acrylamide-co-sodium methacrylate): preparation, swelling and diffusion characteristics evaluation. React Funct Polym 66:1482–1493
Khan KU, Akhtar N, Minhas MU (2020) Poloxamer-407-co-poly (2-acrylamido-2-methylpropane sulfonic acid) cross-linked nanogels for solubility enhancement of olanzapine: synthesis, characterization, and toxicity evaluation. AAPS PharmSciTech. 21:1–15
Ali L, Ahmad M, Usman M, Yousuf M (2014) Controlled release of highly water-soluble antidepressant from hybrid copolymer poly vinyl alcohol hydrogels. Polym Bull 71:31–46
Mutar MA, Radia ND (2012) Controlled release from crosslinked polyacrylic acid as drug delivery theophylline. Iraqi Nat J Chem 45:67–85
Chavda H, Patel C (2011) Effect of crosslinker concentration on characteristics of superporous hydrogel. Int J Pharm Investig 1:17
Swaminathan S, Pastero L, Serpe L, Trotta F, Vavia P, Aquilano D, Trotta M, Zara G, Cavalli R (2010) Cyclodextrin-based nanosponges encapsulating camptothecin: physicochemical characterization, stability and cytotoxicity. Eur J Pharm Biopharm 74:193–201
Ali AE-H, AlArifi A (2009) Characterization and in vitro evaluation of starch based hydrogels as carriers for colon specific drug delivery systems. Carbohydr Polym 78:725–730
Barkat K, Ahmad M, Minhas MU, Khalid I, Malik NS (2020) Chondroitin sulfate-based smart hydrogels for targeted delivery of oxaliplatin in colorectal cancer: preparation, characterization and toxicity evaluation. Polym Bull. 77:6271–6297
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Qaiser, R., Pervaiz, F., Hanan, H. et al. Development of chondroitin sulfate-based mucoadhesive interpenetrating polymeric hydrogels of captopril with adjustable properties as gastro-retentive sustained drug release carriers. Polym. Bull. 81, 5267–5293 (2024). https://doi.org/10.1007/s00289-023-04967-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-023-04967-3