
Abstract
Conventional PMMA bone cement, and more lately BisGMA (bisphenol A-glycidyl methacrylate) composite bone cement, is employed in various bone augmentation procedures such as vertebroplasty. Problems with these materials include high curing exotherm and shrinkage, leakage of toxic components after insertion and exhibition of a modulus mismatch between bone cement and weak bone. A novel high molecular weight dimethacrylate, polypropylene glycol dimethacrylate (PPG), was used in combination with urethane dimethacrylate, hydroxyethyl methacrylate, silica glass particles and fibres to create PPG fibre composite (PFC) dual paste materials. This study was designed to ascertain whether PFCs are a viable alternative to Cortoss™ composite bone void filler and Simplex P™ PMMA bone cement for osteoporotic vertebroplasty and fracture fixation applications. The degree of monomer conversion and curing kinetics of the PFCs and commercial bone cements were found using FTIR, while the mechanical properties were found through biaxial flexural testing. An equation was derived to describe the curing profiles of the PFCs and commercial bone cements. The curing profiles and equations and mechanical properties of the PFCs, Cortoss™ and Simplex P™ bone cements were compared. It was found that PFC materials had more complete monomer conversion, and faster cure than Cortoss™ and Simplex P. The flexural strength of some of the PFC materials was comparable to Cortoss™ and Simplex P. Incorporating fibres into the PFC materials prevented brittle fracture exhibited by Cortoss™ and mimicking the fracture behaviour of Simplex P.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Osteoporosis and osteoarthritis in the increasing elderly population are escalating vertebral fracture incidence and hip and knee implant requirement [1]. Bone fractures and implants are commonly stabilized with polymethyl methacrylate (PMMA) cements [2]. These contain methyl methacrylate (MMA) monomer liquid, a highly reactive, volatile, flammable monomer that has shown high cell toxicity [3, 4] and pre-polymerized PMMA powder [5]. There are many potential concerns with PMMA cements, including property variability [6] and possible contamination [7] from surgeon mixing, less than 100% methacrylate conversion and material shrinkage and poor mechanical properties. After mixing, PMMA cements show a gradual change in viscosity as the polymer beads swell with the monomer [5]. If the cement is placed too soon, cement/ monomer leakage occurs from the site of application. Conversely, if the viscosity becomes too high before placement, bone penetration and micromechanical bonding may be jeopardized [5]. The toxic uncured monomer can leach into surrounding tissues, and high MMA curing exotherm can cause tissue necrosis and fibrosis [6, 8], and polymerization shrinkage can affect the bonding with bone. Mechanical properties of PMMA cement and fracture behaviour limit application, particularly in osteoporotic bone [3, 9].
BisGMA-based Cortoss ™ composite is currently used as an alternative to PMMA cements for fixation of vertebral fractures by cement injection. It contains a mixture of larger, less toxic/volatile dimethacrylate monomers such as BisGMA and TEGDMA (triethylene glycol dimethacrylate) and silica fillers more commonly found in dental composites [10]. A major advantage of Cortoss™ is that it is supplied in double-barreled syringes with automatic mixing tips. This reduces mixing time/variability and reduces contamination risk. Unlike the PMMA beads, the silica fillers do not swell with time. The viscosity of Cortoss™, therefore, remains stable until methacrylate conversion. In contrast to the monomethacrylate monomer in PMMA, Cortoss™ has dimethacrylate monomers that could decrease the leakage of monomer/ cement from the cement mantle after insertion.
Cortoss™, however, is not without drawbacks. The conversion of BisGMA-based composites can be low due to the viscosity of this monomer. High viscosity BisGMA, alone, would also produce a composite that would be incompatible with a mixing syringe. Low conversion and high viscosity are partially overcome through the addition of lower molecular weight TEGDMA but this increases shrinkage and heat generation. Furthermore, Cortoss™ has been shown to exhibit lower or comparable strength and higher modulus than PMMA cements [11,12,13]. The mechanical properties of dental composites have previously been improved by changing the monomer phase and optimizing the filler particle size, size distribution and shape, e.g. fibre addition [14].
It is the hypothesis of this study that the above problems with Cortoss™ may be mitigated through the replacement of BisGMA and TEGDMA with lower viscosity and higher conversion urethane dimethacrylate (UDMA) (10) and higher molecular weight, poly (propylene glycol) dimethacrylate (PPG), respectively, and replacing part of the particulate filler powder with silane-coated glass fibres. The resulting novel PPG fibre composites (PFCs) are described in the following study. The handling, monomer conversion profiles, calculated shrinkage/heat generation and mechanical properties (biaxial flexural strength, stiffness and fracture behaviour) of these PFCs are compared with commercial, “medium viscosity” Simplex P PMMA cement and Cortoss™.
Materials and methods
Materials, variables and mixing
Poly(propylene) Glycol 425 Dimethacrylate (PPG) (Polysciences Inc) was combined with urethane dimethacrylate (UDMA, Esschem plc) in a weight ratio of 3:1, 1:1 or 1:3. UDMA and PPG contained approximately 150 and 100 ppm monomethyl ether hydroquinone (MEHQ) inhibitor, respectively. In addition, UDMA and PPG were supplemented with 70 and 170 ppm butylated hydroxyl toluene (BHT). 7 wt% hydroxyethyl methacrylate (HEMA, Esschem plc) was then added to the PPG-UDMA monomer system to make the PPG-UDMA-HEMA stock monomer. 0.5 wt% of benzoyl peroxide (BP, Polysciences Inc) and 0.5 wt% dimethyl p toluidine (DMPT, Sigma-Aldrich) were added to the stock monomer to make the “initiator and activator monomer”, respectively. The different monomers were mixed using a magnetic stirrer at a speed of 100 rpm for 15 min.
1, 5 or 25 wt% silane-coupled silica glass fibres (GL0271F, d50 15 × 300 μm from Mosci) were mixed with silane-coupled radiopaque silica glass filler (IF2019, d99 40 μm, d80 30 μm, d60 10 μm from SciPharm). The resultant powders phase (60, 75 or 80 wt%) were hand mixed with the initiator and activator monomers to make separate initiator and activator pastes (Fig. 1).

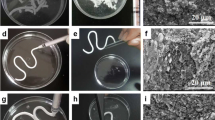
Outline of steps in the sample preparation process. a Initiator (BP) or activator (DMPT) is dissolved in the monomer phase which is subsequently mixed by hand with particulate and fibre fillers. b Each barrel of a 10 cc double-barrelled syringe is filled with either initiator or an activator containing paste. c Two pastes are mixed using the double-barrelled syringe, mixing tip and delivery gun. d 10-mm diameter, 1-mm-thick disc specimen after cure, but before 24-h hydration, is removed from brass split ring mould and any excess material cut away
Eight different formulations were investigated with three variables (PPG, powder (P) or fibre (F) content) using a two-level factorial design with one intermediate formulation (Table 1). The separate “initiator” and “activator” pastes, for a given formulation, were loaded into opposite sides of a double-barrel syringe (Sulzer Mixpac) with 2 × 5 cm3 barrels. Equal volumes of the initiator and activator pastes could then be rapidly combined, by expulsion through the syringe automatic mixing tips, as required (Fig. 1).
Simplex P PMMA bone cement (Stryker) powder consists of 29.4 g PMMA–styrene (molecular weight 306,000 g mol−1), 6-g pre-polymerized PMMA beads (molecular weight 974,000 g mol−1), 4-g barium sulphate and 0.6-g BP. The liquid contains 19.5-ml MMA, 0.5-ml DMPT and 80-ppm hydroquinone (HQ) [15]. PMMA cement was mixed as per package instructions.
Cortoss™ (Styker) consists of the dimethacrylate monomers combined with 69 wt% inorganic glass–ceramic filler (5–50 μm), coupled with 2.4 wt% 3-methylacryloxypropyltrimethoxysilane [11] and the bioactive glass–ceramic filler combeite [16]. The Cortoss™ bone void filler was mixed via delivery gun and mixing tips provided by the manufacturer and cured via chemically initiated (benzoyl peroxide and DHEPT) free radical polymerization. The methods of commercial specimen manufacture were the same as those prepared from the PPG composite materials.
Polymerization setting reaction and shrinkage
Material FTIR spectra were obtained every 4 s for 20 min using a series 2000 FTIR spectrometer (PerkinElmer) with a golden gate diamond ATR attachment (Specac) and Timebase software (at 25 ℃). Brass rings (1-mm thick, 10-mm diameter) were placed around the ATR diamond and were filled with the mixed paste directly from the double-barrel syringes or with Simplex P after 1 min of mixing. The top surface of the mixed paste was covered with an acetate sheet to prevent any interference with the polymerization reaction.
Difference spectra were obtained to ensure methacrylate polymerization was the only process causing spectral changes [12]. Fractional reaction extent (ξ) is calculated using Eq. 1 [13].
A0, At and Af represent 1320 cm− 1 absorbance values at initial time, at time t, and at an extrapolated final time (determined by plotting data versus inverse time), respectively. The reaction half-life (t0.5) was taken as the time at which the reaction extent was 50% of its final level. Reaction extent was plotted against time divided by t0.5 to address if methacrylate conversion profiles could be described by a single equation [17]. Monomer conversion was determined from the height of the 1320 cm−1 peak above the background at 1350 cm−1 (Fig. 2). Furthermore, polymerization shrinkage was estimated from known compositions, and extrapolated final conversions assuming one mole of polymerizing C=C bonds give volumetric shrinkage of 23 cm3 [18].

FTIR spectra of curing PFC5 with time. Inset shows the change in absorbance used to determine the cure profile. The 1320 cm−1 peak used to measure peak height corresponds to C–O bond in the polymerizing methacrylate group. Only peaks corresponding to the methacrylate group are changing—indicating only one reaction (polymerization) is proceeding
Biaxial flexural strength and Young’s Modulus
For mechanical testing, disc specimens were prepared in a dimension of 10 mm in diameter and 1 mm in thickness using split brass rings. Specimens were left for 24 h, at 25 °C, before extraction from the rings. Any excess material, around the edges, was removed to make a smooth edge (Fig. 1). Prior to mechanical testing, cured specimens were stored in 10 ml of deionized water and were incubated at 37 °C for 24 h.
A “Ball on Ring” biaxial testing method was employed as reported previously by Palin and Mehdawi [19, 20]. Each specimen was placed on a knife-edge ring support (radius a = 4 mm) and then loaded using a spherical tip in an Instron 4505 Universal Testing Machine. Biaxial flexural strength (BFS) was determined using Eq. 2 [21].
where Pmax maximum load (N), d average thickness of the sample (mm), a support radius (mm), ν Poisson’s ratio (estimated as 0.3 due to the strain rate employed in the biaxial test method) [14].
Elastic modulus, E, was calculated from the maximum initial slope of load versus central deflection (dP/dɷ) using Eq. 3 [22].
Statistical analysis
The data will be presented in form of a mean ± 95% confidence interval (CI). n = 4 for FTIR studies and 8 for mechanical data. Property values are significantly different if error bars do not overlap. SPSS v 21 was used to analyse the monomer conversion, half-life and polymerization shrinkage. One-way ANOVA followed by post hoc Tukey’s test was used to analyse the data. The p-value of less than equal to ( ≤)0.01 will be considered significant.
Results
Sample handling and syringe use
For all PFCs, initial viscosity was sufficiently low to enable the use of a commercial syringe and automatic mixing tips. It took between 2 and 4 s for the pastes to flow through the mixing tips. The pressure on the gun required was higher for formulations with greater powder content and lower PPG due to their greater viscosity. While the syringe was in continuous use, the paste did not set in the tip. Material left within the tip set between two and seven minutes. The viscosity of the Cortoss™ paste was similar to PFCs containing high filler content and low level of PPG (PFC 3 and 4). The bore of the Cortoss™ mixing tip was larger than those used for PFCs, and so the pressure required extruding the Cortoss™ was similar to that required for PFC 7 and 8 but lower than for PFC 3 and 4. Simplex P took one minute to mix by hand and 10 min to set at room temperature.
Setting and shrinkage
FTIR spectra of curing PFCs and Cortoss™ were similar to those of polymerizing dental composites (Fig. 2). Difference spectra were characteristic of methacrylate polymerization. These had maxima at 1250 cm−1 and minima at 1300 and 1320 cm−1 due to a change in methacrylate C–O vibration as tabulated in Table 2. They also indicated change at 1700 and 1640 cm−1 due to methacrylate C=O vibrational shifts and C=C loss, respectively (data not shown). With Simplex P, there were small changes in the first few minutes characteristic of dissolution of polymer into the monomer phase before changes characteristic of methacrylate polymerization.
Examples of reaction extent versus time divided by t0.5 are provided in Fig. 3. All materials demonstrated an inhibition period followed by a rapid reaction. By 4t0.5, reaction extent was over 80% for all materials. Below t/t0.5 = ti = 0.35 ± 0.04, reaction extent was negligible for all PFCs Fig. 3. Conversely, in the range 0.35 < t/t0.5 < 1, a constant gradient (dζ/d(t/t0.5) = 0.5/(1 − ti) = (0.77 ± 0.03)) was observed. In this range, reaction extent was therefore given by

Reaction extent, Ϛ, (see Eq. 1) versus normalized time, t/t0.5. Within experimental error, this plot for all formulations overlaps when Ϛ < 50%. Beyond this point reaction extent versus t/t0.5 tends to 1 more rapidly if the PPG level is high. The legend provides formulation composition in order of reaction extent at t/t0.5 = 3
For Cortoss™ and Simplex P, similar equations could describe the results but with ti = 0.52 and 0.77, respectively. t0.5 values for all formulations are provided in Table 1 and Fig. 4. Increased PPG and powder content both significantly raised PFC t0.5 when fibre content is held constant. With 60% powder, t0.5 (averaged over fibre content) are 72 s and 184 s with 23 and 70% PPG, respectively. With 80% powder, these increased to 132 s and 246 s. Cortoss™ t0.5 (231 s) was similar to that of PFC 5–8. Simplex P t0.5 (403 s) was higher than that of all other materials.

Monomer conversion, time to half-reaction and polymerization shrinkage of commercial formulations (Cortoss and Simplex P) and experimental PFC formulations are presented. Low filler, low PPG denotes a formulation containing 60 wt% filler and 23% PPG monomer as described in the methods section, filled/open symbols denote high and low fibre content, respectively. Formulations containing 70% PPG show significantly higher monomer conversion in comparison with the 23% PPG formulations, Cortoss and Simplex P. PFCs containing the same monomer mix exhibit conversion percentages that are not significantly different, irrespective of filler and fibre content. When considering time to half-reaction, values for high-fibre PFCs are not significantly different to those with low fibre content when overall filler content is low. Shrinkage of Cortoss is not significantly different to shrinkage of high PPG, and high filler PFC. In addition, Simplex P does not have significantly different polymerization shrinkage when compared with formulations containing 70% PPG and low filler levels. All for p ≥ 0.01. (Errors bars: 95% CI, n = 4)
PPG content was the only variable to significantly increase PFC final conversion (p ≤ 0.01). It increased on average from 70 to 97% with an increasing PPG level from 23 to 70 wt% of the monomer. Monomer conversion of PFC 4 (73%) was close to the average result and that of Simplex P (76%). Conversion of Cortoss™ was the lowest (64%). Calculated shrinkages of PFCs with high powder content were comparable to or lower than that of Cortoss™ (3.7%) and Simplex (6.2%), respectively (see Table 1).
Biaxial flexure strength, Young's Modulus and fracture behaviour
All disc specimens with low fibre content (1%) had a brittle failure. Raising fibre content had no significant effect on load or deflection at yield but increased deflection at final failure. Load versus displacement of Simplex P PMMA was similar to that of PFC 2.
PFC BFS ranged between 37 and 158 MPa and was affected most by PPG level (Fig. 5). Raising powder content increased strength at high PPG levels but had the opposite effect at low PPG. The strength of Cortoss™ (120 MPa) and Simplex P (131 MPa) were comparable with the average result for PFC with low PPG content.

Average BFS and modulus for varying PPG percentage in monomer (PPG) and total powder percentage (P). When the level of PPG is high, raising the powder content increases strength. At low PPG levels, increasing powder content does not increase strength. Error bars are 95% confidence interval (n = 16, averaged over 8 high fibre content, and 8 low fibre specimens as fibre content did not affect BFS or modulus)
PFC moduli ranged between 0.3 and 2.9 GPa and were reduced most by increasing PPG. It also declined, however, on reducing powder content (Fig. 5). The modulus of PFC 1 (1.8 GPa) was comparable with that of Simplex P (1.6 GPa). Cortoss™’s stiffness (3 GPa) was found to be significantly higher than all but PFC 3 and 4.
Discussion
Handling
The glass particles and fibres used in the above study are silane-treated enabling greater filler content [23, 24]. The highest PFC filler content of 80 wt% was 11 wt% higher than in Cortoss™. Zhang et al. have found that fibre incorporation affected the handling of methacrylate composites by increasing the viscosity [25]. In the present study, the silane treatment and aspect ratio of incorporated fibres were such that the handling of the material was in no way compromised.
The easy and rapid PFC mixing would save the surgeon time and facilitate a fast set. Application via the mixing tip end directly into the implant site would avoid contact with other operating theatre surfaces and thereby reduce bacterial contamination risk.
Setting and shrinkage
The mechanism of free radical polymerization of dimethacrylates involves initiation, inhibition, propagation, crosslinking and termination steps. After mixing of initiator and activator pastes, the rate of the polymerization initiation, Ri, and concentration of inhibitor [Z] at a time, t, will be given by [26,27,28]:
\(R_{i} \, = \,2fk_{d} \left[ A \right]\left[ B \right]\) and \(\left[ Z \right]\, = \,\left[ {Z_{o} } \right]\, - \,R_{i} t\) kd is the rate constant for initiation, f is the initiator efficiency, and [A], [B] and [Z0] are the amine, BP and initial inhibitor concentrations in the monomer. If all the inhibitors must be consumed prior to any monomer polymerization, the above equations predict an inhibition period given by
In the above PFC formulations, amine and initiator concentrations are constant. The approximate doubling of inhibition time with raising PPG content may therefore be a consequence of varying inhibitor concentrations and/or fkd being larger for UDMA than PPG. The increased inhibition time with the highest powder content may be due to greater oxygen incorporation due to filler not being completely “wet” by the monomer increasing oxygen inhibition.
Higher ti of Cortoss™ is likely due to a combination of different monomers and inhibitors. Simplex P contains double the concentrations of BP and DMPT with fewer inhibitors. The most likely reason for greater reaction delay and higher ti is therefore that BP must dissolve/diffuse, from the surface of the filler phase, into the monomer phase before any reaction can occur.
Immediately subsequent to the inhibition period, the polymer concentration will be negligible. Furthermore, the total number of polymer molecules formed is much smaller than the initial monomer molecules. Moreover, larger polymer molecules diffuse more slowly. Most of the dimethacrylate molecules will therefore likely form into linear chains with pendant unreacted methacrylate groups before any crosslinking. From Eq. 4, in the experimental PFCs, the rate of this process is given by:
− d[M]/dt is the average number of methacrylate groups per monomer and C final conversion. As C and t0.5 increase on average by factors of 1.3 and 1.9, the rate of early monomer reaction therefore decreases by a factor of 1.5 on raising PPG level from its low to high percentage. This could be a consequence of fkd being larger for UDMA than PPG as the initial rate of a monomer polymerization is proportional to the square root of Ri [26,27,28]. Altering the monomer could also, however, be affecting the rate constants for propagation and termination steps.
As the reaction proceeds, the rate should decline due to monomer consumption but increase due to the “gel effect” [27] (31). More viscous monomers, such as BisGMA and UDMA, have a more pronounced auto-acceleration phase (due to the gel effect) than less viscous monomers, such as TEG DMA or PPG [28] or methyl methacrylate. The higher concentrations of initiators and low inhibitor levels in Simplex P would enable rapid reaction once the BP has dissolved. Fast reaction, however, may lower mechanical properties due to a reduction in polymer molecular weight. Furthermore, it may cause greater temperature rise due to reduced time for heat dissipation.
After 50% reaction, methacrylate conversion for the dimethacrylate containing PFC’s and Cortoss™ declined due to the slower crosslinking process. As final conversions are much higher than 50%, in these materials there is unlikely to be significant unbound monomer to cause toxicity concerns. Conversely, with Simplex, 100% conversion is required to ensure no free monomer. Polymerization generally ceases when material Tg is 20–40 ℃ above the surrounding temperature [29]. As monomers are consumed, Tg increases [30]. Crosslinking and/or increase in polymer molecular weight also increases Tg [30] but to a lesser extent. The conversion of Cortoss™ and the PFCs with low PPGDMA observed above are comparable with those typically obtained with BISGMA/ UDMA/ TEGDMA-based polymers and composites [30]. The greater chain flexibility and lower Tg of polymerized PPG would explain the increased crosslinking upon raising the level of this component.
Fully cured PMMA has a Tg of ~ 107 ℃ [31] preventing conversion from reaching 100% at 25 ℃. In the body, however, the use of larger volumes of cement will lead to high-temperature rises. Although this may cause tissue necrosis, it will enable greater conversion. The PMMA conversion ceases more abruptly than observed with the PFC; this would be consistent with Simplex P not having a slower crosslinking reaction. The level of heat generation will be proportional to the shrinkage. In the above PFCs, this is kept low by raising the filler content and reducing polymerization. This would potentially aid in reducing the thermal necrosis of the bone tissue.
To improve bone cement hydrophilicity and better bone wetting, HEMA has been added in this study. This hydrophilicity of HEMA is characterized by high water sorption, which in turn can compensate for the shrinkage that happens during the polymerization reaction. However, it should be added in a smaller amount as it can induce higher expansion of bone cement and can a higher failure of the cement.
Biaxial flexural strength, modulus and fracture behaviour
In flexural strength studies, the sample is under compression on the top surface and tension on the lower surface. The latter generally has the most influence on flexural strength; with crystalline materials, maximum theoretical tensile strength is proportional to modulus but this is reduced by flaws that initiate crack propagation [27]. In amorphous polymers such as PMMA, strength is increased by raising molecular weight and chain entanglements. Crosslinking, however, prevents polymers from disentangling. In a network with a high level of crosslinking, C–C covalent bonds must be broken for the material to fail.
In composite materials, the interface between phases is often the point of greatest weakness/cause of failure. Silane treatment of filler enables chemical bonding between the monomer and filler phases during polymerization [32]. In PFCs and Cortoss™, silane treatment of the filler increases monomer wetting enabling strength comparable to PMMA. With PMMA, a long period of dissolution prior to cure may reduce interfacial weakness at the bead/monomer interface. Preventing flocculation of the radiopacifying components (e.g. barium sulphate) could also improve strength. In PFCs containing high levels of PPG, increasing filler content raised strength. In this case, the monomer is very fluid which enables good particle wetting. After cure, the load may then be more effectively transferred to stronger filler particles. With low PPG, strength declined with raising powder content. This may be due to the polymer phase being stronger than the glass. Alternatively, the more viscous monomer may less effectively wet the filler particles. Particle agglomerates may then form particularly at high powder content. Agglomerates reduce strength by introducing flaws [25]. They may also trap oxygen locally reducing chemical and physical bonding between the filler and organic phase [33].
High aspect ratio fibres can provide crack control behaviour that is dependent on the matrix properties and interfacial bond strength [34]. Brittle fracture of the PFC with low fibre content was typical of that observed with conventional dental composites and Cortoss™ [35]. With high fibre content, the load was carried, with increasing deflection up until the fibres failed. This fracture behaviour is more comparable with that of Simplex than Cortoss™.
For traditional engineering materials, like titanium, strength is usually of the order of 2% of Young’s modulus [36]. Composite bone cements, such as Cortoss™ have strength of around 4% of the modulus. For Simplex P cement, this value is higher, at around 8%. For PFCs, the extension at failure is larger still and the strength can reach up to 12% of the stiffness. Reducing the modulus relative to the strength is particularly important in applications such as vertebroplasty and screw augmentation when stabilizing osteoporotic hip fractures. More flexible cement materials may prevent damage to adjacent vertebrae, as over stiffening of cement-treated vertebrae can be a major cause of adjacent vertebral fracture [37].
Conclusions
PPG has been used in combination with UDMA, HEMA, silica glass particles and fibres to create PPG fibre composite (PFC) dual paste materials of bone cement with rheological characteristics suitable for use in a commercial double-barrelled syringe with a smaller bore mixing tip than Cortoss™. Handling properties were improved with the use of a double-barrel syringe as compared to manual mixing of Simplex cement. Furthermore, the viscosity of the experimental composite was found similar to Cortoss™. This automatic mixing reduces bubble formation during mixing with limited material loss and time. Better experimental composite reaction rate improved the monomer conversion (as shown by FTIR analysis) and lower shrinkage, exhibiting better biocompatibility than Cortoss™ and Simplex™. The strength of PFCs was comparable to Cortoss™ but those with high levels of PPG had lower modulus which better matches that of bone and prevents damage to adjacent vertebrae when used in vertebroplasty. The prepared injectable composite bone repair material would be well-suited to fixation of osteoporotic bone in vertebroplasty and screw augmentation of hip fracture fixation procedures.
References
Van Staa TP, Dennison EM, Leufkens HG, Cooper C (2001) Epidemiology of fractures in England and Wales. Bone 29(6):517–522
Lewis G, Xu H, Madigan S, Towler MR (2007) Influence of strontia on various properties of Surgical Simplex (R) P acrylic bone cement and experimental variants. Acta Biomater 3(6):970–979
Kuehn K-D, Ege W, Gopp U (2005) Acrylic bone cements: composition and properties. Orthop Clin North Am 36(1):17–28
Fries IB, Fisher AA, Salvati EA (1975) Contact dermatitis in surgeons from methylmethacrylate bone cement. J Bone Joint Surg Am 57(4):547–549
Wnek GE (2008) Encyclopedia of biomaterials and biomedical engineering 2nd (eds) , Informa Healthcare, London p 4
Lewis G (1997) Properties of acrylic bone cement: State of the art review. J Biomed Mater Res 38(2):155–182
Jiranek WA, Hanssen AD, Greenwald AS (2006) Antibiotic-loaded bone cement for infection prophylaxis in total joint replacement. J Bone Jt Surg 88(11):2487–2500
Santin M, Motta A, Borzachiello A, Nicolais L, Ambrosio L (2004) Effect of PMMA cement radical polymerisation on the inflammatory response. J Mater Sci Mater Med 15(11):1175–1180
Kane RJ, Yue W, Mason JJ, Roeder RK (2010) Improved fatigue life of acrylic bone cements reinforced with zirconia fibers RID A-9398-2008. J Mech Behav Biomed Mater 3(7):504–511
Lewis A (2009) Drug Device Combination Products Delivery Technologies and Applications. Woodhead Publishing Ltd, Sawston, p 560
Pomrink GJ, DiCicco MP, Clineff TD, Erbe EM (2003) Evaluation of the reaction kinetics of Cortoss, a thermoset cortical bone void filler. Biomater 24(6):1023–1031
Young AM, Rafeeka SA, Howlett JA (2004) FTIR investigation of monomer polymerisation and polyacid neutralisation kinetics and mechanisms in various aesthetic dental restorative materials. Biomater 25(5):823–833
Young AM, Ng PYJ, Gbureck U, Nazhat SN, Barralet JE, Hofmann MP (2008) Characterization of chlorhexidine-releasing, fast-setting, brushite bone cements. Acta Biomater 4(4):1081–1088
Chung S, Yap A, Koh W, Tsai K, Lim C (2004) Measurement of Poisson’s ratio of dental composite restorative materials. Biomater 25(13):2455–2460
Madigan S, Towler MR, Lewis G (2006) Optimisation of the composition of an acrylic bone cement: application to relative amounts of the initiator and the activator/co-initiator in Surgical Simplex P. J Mater Sci Mater Med 17(4):307–311
Sanus GZ, Tanriverdi T, Kafadar AM, Ulu MO, Uzan M (2005) Use of Cortoss for reconstruction of anterior cranial base: a preliminary clinical experience. Eur J Plast Surg 27(8):371–377
Hofmann MP, Young AM, Gbureck U, Nazhat SN, Barralet JE (2006) FTIR-monitoring of a fast setting brushite bone cement: effect of intermediate phases. J Mater Chem 16(31):3199–3206
Rueggeberg F, Tamareselvy K (1995) Resin cure determination by polymerization shrinkage. Dent Mater 11(4):265–268
Palin WM, Fleming GJ, Marquis PM (2005) The reliability of standardized flexure strength testing procedures for a light-activated resin-based composite. Dent Mater 21(10):911–919
Mehdawi I, Abou Neel EA, Valappil SP, Palmer G, Salih V, Pratten J et al (2009) Development of remineralizing, antibacterial dental materials. Acta Biomater 5(7):2525–2539
Timoshenko SP, Kreiger SW (1964) Theory of plates and shells, 2nd edn. McGraw Hill Higher Education, New York
Higgs W, Lucksanasombool P, Higgs R, Swain M (2001) A simple method of determining the modulus of orthopedic bone cement. J Biomed Mater Res 58(2):188–195
Wilson KS, Zhang K, Antonucci JM (2005) Systematic variation of interfacial phase reactivity in dental nanocomposites. Biomater 26(25):5095–5103
Karmaker A, Prasad A, Sarkar N (2007) Characterization of adsorbed silane on fillers used in dental composite restoratives and its effect on composite properties. J Mater Sci Mater Med 18(6):1157–1162
Zhang H, Zhang M (2010) Effect of surface treatment of hydroxyapatite whiskers on the mechanical properties of bis-GMA-based composites. Biomed Mater 5(5):054106
Odian G (2004) Principles of Polymerization, 4th edn. Wiley Interscience, New York
Young RJ, Lovell PA (1991) Introduction to polymers, 2nd edn. CRC Press, Boca Raton
Sideridou ID, Achilias DS, Kostidou NC (2008) Copolymerization kinetics of dental dimethacrylate resins initiated by a benzoyl peroxide/amine redox system. J Appl Polym Sci 109(1):515–524
Turi EA (1997) Thermal characterization of polymeric materials. Academic Press, New York, p 1200
Sideridou I, Tserki V, Papanastasiou G (2002) Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomater 23(8):1819–1829
Abou Neel EA, Salih V, Revell PA, Young AM (2012) Viscoelastic and biological performance of low-modulus, reactive calcium phosphate-filled, degradable, polymeric bone adhesives. Acta Biomater 8(1):313–320
Tacir I, Kama J, Zortuk M, Eskimez S (2006) Flexural properties of glass fibre reinforced acrylic resin polymers. Aust Dent J 51(1):52–56
Vallittu P (1997) Oxygen inhibition of autopolymerization of polymethyl methacrylate-glass fibre composite. J Mater Sci Mater Med 8(8):489–492
Garoushi S, Vallittu P, Lassila L (2007) Short glass fiber reinforced restorative composite resin with semi-inter penetrating polymer network matrix. Dental Mater 23(11):1356–1362
Chung SM, Yap AUJ, Chandra SP, Lim CT (2004) Flexural strength of dental composite restoratives: comparison of biaxial and three-point bending test. J Biomed Mater Res Part B Appl Biomater 71B(2):278–283
Shi L, Shi L, Wang L, Duan Y, Lei W, Wang Z et al (2013) The improved biological performance of a novel low elastic modulus implant. PLoS ONE 8(2):e55015
Belkoff S, Mathis J, Erbe E, Fenton D (2000) Biomechanical evaluation of a new bone cement for use in vertebroplasty. Spine 25(9):1061–1064
Acknowledgements
This work has been supported by UCL Eastman Dental Institute, 256 Gray’s Inn Road, London WC1X 8LD, UK
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Main, K., Khan, M.A., Nuutinen, JP. et al. Evaluation of modified dental composites as an alternative to Poly(methyl methacrylate) bone cement. Polym. Bull. 80, 13143–13158 (2023). https://doi.org/10.1007/s00289-023-04677-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-023-04677-w