
Abstract
A series of polyethylene terephthalate-co-[(6-oxido-6H-dibenz[c,e][1,2]oxaphosphorin-6-yl)methyl] butanedioate (PET-co-PEDDP) copolyesters were prepared through the reaction among ethylene glycol (EG), terephthalic acid, and [(6-oxido-6H-dibenz[c,e] oxaphosphorin-6-yl)methyl]butanedioic acid (DDP) through the direct esterification and polycondensation processes. Structure study of the prepared copolyesters using Fourier transform infrared spectroscopy and nuclear magnetic resonance (NMR) indicates polyester production. It is concluded that DDP comonomer incorporated into the polymer chain. Examination of the thermal treatment of all samples detected no phase separation. Melting point linearly decreases with DDP content; the Baur’s equation could describe the melting point. Melt crystallization kinetics of all synthesized samples in a wide range of cooling rates were investigated by various kinetics models. Due to secondary crystallization, some simple models such as the Avrami equation could not fit the data very well. The best crystallization kinetics model was the Hay model by taking into consideration the secondary crystallization, which is the main achievement of this research. Furthermore, the effects of comonomer on the thermal degradation of copolyesters were also analyzed using thermal gravimetry analysis. The Coats–Redfern equation was applied to examine the influence of comonomer on thermal degradation.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polyethylene terephthalate (PET) is the most significant and generated member of polyesters that are a type of polymer containing ester functional groups in their backbone chain. The superior chemical and mechanical features of PET have made it applicable in various industrial applications such as fiber, textile, film, and packaging [1, 2]. The global PET production in 2024 will be approximately 35 million metric tons [3]. Nevertheless, there are still several drawbacks for PET utilization, such as its poor processability, crystallinity, and flame behavior. PET could be categorized in oxygen-containing polymers [4]. Various approaches could overcome the mentioned shortages, including adding fillers and additives, polymer blending, and copolymerization. However, such manipulations affect crystallization behavior which is important from practical point of view in final article production.
Ibbotson and Sheldon studied the effect of various additives on the crystallization behavior of PET [5]. They used DSC under dynamic and isothermal modes. Phang et al. prepared polyester and nanoclay composites via melt compounding [6]. They study was based on PET and its copolyetser, supplied by Eastman Co., nanocomposites. Mohsen-Nia and Memarzadeh prepared poly(ethylene sebacate) (PESeb) and PESeb/silica nanocomposites by in situ polymerization [7]. They studied non-isothermal crystallization behavior of PESeb/SiO2 nanocomposites and applied various theoretical equations such as Avrami, Ozawa, and combined Avrami and Ozawa. It was reported that nanoparticles of SiO2 influenced the mechanism of nucleation and the growth of crystallites. Kahkesh and Rafizadeh prepared poly(butylene succinate)/nano-boehmite composites with an in situ polymerization to examine their crystallization, thermal degradation, and flame retardancy behaviors [8]. Agrawal et al. presented non-isothermal kinetics of PET-titanium dioxide (TiO2) nanocomposites [9]. They explained this kinetics through Avrami–Ozawa combined theory. Lin et al. investigated kinetics of pure PET, PET/mica, and PET/TiO2-coated mica composites using DSC [10]. They examined modified Avrami, Ozawa, and Mo methods. Moreover, they calculated activation energies of non-isothermal crystallization by Kissinger and Flynn–Wall–Ozawa methods. Gao et al. introduced PET/hydrophobic BaSO4 nanocomposites via in situ polymerization [11]. Their observations exhibited an improvement in the PET stability caused by BaSO4 addition and an increment of crystallization rate. Valapa et al. prepared poly(lactic acid) (PLA)/sucrose palmitate (SP) nanocomposites by solution casting method [12]. They evaluated Avrami model parameters for cold crystallization kinetics. Wang et al. used DSC and POM analyses to evaluate the isothermal crystallization of neat PET and its nanocomposites with NaMMT, prepared by solid-state mechanochemical and conventional methods [13].
Polymer’s molecular weight and structure, like branching, have significant influences on its properties. Mohammadi et al. surveyed the impact of initial crystallization conditions on solid-state polymerization of PET and molecular weight of products [14]. They also checked out the effect of molecular weight on PET crystallization. Papageorgiou et al. studied the crystallization kinetics of branched and partially crosslinked PET using Wide-Angle X-ray Diffraction (WAXD) and DSC analyses [15]. Their samples were produced by trimethyl trimellitate as a branching agent. The content of the branching agent enhances the isothermal crystallization half-times. Saeed et al. produced blends of recycled PET and hyperbranched polyester (HBPET) [16]. They analyzed kinetics of non-isothermal crystallization with modified Avrami and Ozawa equations and Mo method. Then, the Kissinger method was applied to determine activation energy of crystallization of RPET. Ravari et al. studied effect of L-lactide dimer on the crystallization of poly(L-lactide) (PLLA) [17]. They conducted the non-isothermal crystallization kinetics using Avrami, Jeziorny, Ozawa, and Mo. Wang et al. synthesized a copolymer of PET and bis 4-carboxyphenyl phenyl phosphine oxide (BCPPO), illustrating an improved flame retarding behavior [18]. By determining the transition points of Tg and Tm by DSC analysis, they detected a decrease in Tm and an increase in Tg. Wang et al. investigated the thermal performance of a series of copolyesters containing phosphorus using DSC and Thermal Gravimetric (TGA) analyses [19]. They fabricated the samples with the direct esterification of [(6-oxido-6H-dibenz (c,e) [1,2] oxaphosphorin-6-yl) methyl]-butanedioic acid (ODOP-BDA) and bis (hydrocyethyl) terephthalate (BHET) or bis (hydroxyethyl) naphthalate (BHEN). They deduced that both Tg and Tm are reduced by increasing the comonomer content. Cheng and Cheng synthesized a PET-co-poly(ethylene DDP)s copolyester and exhibited that higher phosphorus content causes a reduction in the crystallinity and melting temperature [20]. Avarzman et al. synthesized branched polyester with terephthalic acid (TPA), isophthalic acid (IPA), and trimellitic anhydride (TMA) as comonomers [21]. They reported structure and crystallization behavior. Zhou et al. synthesized poly(butylene succinate) (PBS)/9,10-dihydro-10-[2,3-di(hydroxycarbonyl)propyl]-10-phosphaphenanthrene-10-oxide (DDP) copolyesters containing phosphorus pendent group [22]. They explored the composition, crystallization behavior, multiple melting behavior, and spherulitic morphology of the copolyesters. Naghavi et al. synthesized short-segmented block copolymers of poly(butylene succinate-co-butylene fumarate) (PBS-BF) and computed the Avrami equation parameters using nonlinear Levenberg–Marquardt algorithm [23]. Al-Mulla presented DSC results of non-isothermal cooling of polytrimethylene terephthalate (PTT)/polybutylene terephthalate (PBT)/and polycarbonate (PC) blend [24]. He evaluated the kinetic parameters of crystallization based on the Kissinger and Matusita models. Kint and Muñoz‐Guerra reviewed publications that had been related to the thermally induced crystallization of PET and its copolyesters [25]. They included some illustrative data for isothermal and non-isothermal crystallization. Chen et al. produced poly[(trimethylene terephthalate)-co-(38 mol% ethylene terephthalate)] copolyester [26]. Then, crystallization kinetics and melting behavior of copolyesters were presented.
Based on the literature review, there have been many published data on crystallization kinetics for various polymers and polyesters. Moreover, the performance of polymers, especially PET, could be manipulated by their thermal behavior, degree of crystallinity, and other features. In the current work, a series of copolyesters consisting of ethylene glycol, terephthalic acid, and [(6-oxido-6H-dibenz[c,e][1,2] oxaphosphorin-6-yl) methyl] butanedioic acid (DDP), as a phosphorous-containing comonomer, are synthesized. The samples have been characterized by various analyses such as Fourier Transform Infrared Spectroscopy (FTIR), proton nuclear magnetic resonance (HNMR, and TGA. Furthermore, the thermal behavior and crystallization are examined by non-isothermal DSC analysis in various cooling rates (melt crystallization) and one heating rate (cold crystallization).
Materials and methods
Materials
Ethylene glycol (EG) and terephthalic acid (TPA) as main monomers were supplied by Shahid Tondgooyan Petrochemical Company (STPC). [(6-oxido-6H-dibenz[c,e] oxaphosphorin-6-yl)methyl]butanedioic acid (DDP), as comonomers, was purchased from Phrmicell Chemical Business Unit, South Korea. Antimony oxide, as the polycondensation catalyst, was prepared by Polychem Industrial Co., Ltd., China. Di chloroacetic acid, as the solvent in the intrinsic viscosity measurement, orthocresol and chloroform, as the solvents in acid end group measurement, and potassium hydroxide, as the agent in potentiometry, were bought from Merck Co., Germany.
Synthesis of polyethylene terephthalate and its copolyesters
A homemade laboratory-scale reactor was employed to generate all samples. Figure 1 displays the scheme of the used experimental setup. As can be observed, it consists of a one-liter stainless steel reactor, EG condenser, water separator, heating system, and vacuum pump as main constituents of the system.

Schematic view of experimental setup for synthesizing PET
Table 1 exhibits the compositions of the produced samples, DDP, and phosphorus element contents at different analyses. Some preliminary runs were carried out to specify the proper process variables for getting the appropriate intrinsic viscosity as an implication of the molecular weight. Every batch was conducted in such a way as to obtain about 550 g of polyester. Afterward, the amount of 0.19 g Sb2O3 was added to the system. The OH to COOH mole ratio was assumed fixed at 1.4 in all runs.
The weight percent of DDP in the final polymer was selected to be up to 11. This range of DDP content is in such a way that be economical in the copolyester production from the commercial point of view. Moreover, this amount of comonomer may affect some properties such as flame retardancy, while keeping PET properties. The weighted raw materials were agitated for 20 min under standard laboratory conditions to get a uniform paste. Afterward, the paste was poured into the reactor and completely sealed. The EG condenser temperature was set to 160 °C during the course of the esterification step. The mixture temperature was reached above 195 °C under 2 bar pressure using a heating system and then mixed for about 10 min. Subsequently, the mixture temperature and pressure were increased to 255 °C and 5 bar, respectively. The water production testified as to the start of the esterification step. The distilled water gathered every 15 min as an indication of the esterification reaction progress. The termination of water production determines the end of the esterification step. Accordingly, the mixture temperature increased to 275 °C, and a vacuum was applied to ease EG removal and polycondensation progress. After 120 min, the synthesized polyester was evacuated from the reactor. Besides that, the mixing rate is 100 rpm over the whole course of synthesis.
Characterization of samples
The intrinsic viscosity (IV) of samples was measured by the solution viscometry based on the ISO-1628-5 standard. The sample dissolved in the dichloroacetic acid with a concentration of 10 mg/mL under a temperature range of 85–100 °C. The IV was determined by a Lauda–Konigshofen viscometer at 25 ± 0.1 °C. The acid end groups were specified using the gravimetric approach. 1 g of sample poured in a 70:30 mixture of orthocresol: chloroform under heating and reflux. Then, the solution was titrated with 0.5 M KOH solution in the ethanol with a model 798 MPT Titrino Metrrohm titrator with Solvotrode model electrode. The end of titration is designated by the potentiometry and Tiamo 2.4 software. The acid group concentration at the end of the esterification step is called acid number AN.
To measure diethylene glycol (DEG), sample was gone under transesterification in methanol. The amount of 1–1.5 g sample was poured into 30 mL of transesterification solution, consisting of 2.0 g tetraethylene glycol dimethyl ether, 300 mg zinc acetate, and methanol, heated to 220 °C, and then kept for two hours to measure the content of DEG. Consequently, the final solution cooled to room temperature, and dimethyl terephthalate crystals were filtered. The remained solution was injected into gas chromatography, GC3800 model of Varian Co., to determine the DEG content.
The parameters of L, i.e., transparency, and b, i.e., yellowness, as color indices, have been measured by a BYK spectrophotometer. Amorphous samples have been crystallized for 30 min at 150 °C before performing the test. FTIR spectra were yielded with a Bruker FTIR spectrometer at room temperature. The amount of 5–10 mg sample was powdered and mixed with KBr to form a pellet. HNMR spectrometry performed using a Bruker 400 MHz UltraShield branded instrument. The sample dissolved into trifluoroacetic acid, and a little amount of CDCl3 was added for locking the detector.
A Swiss-made Metller Toledo, 822e model, was used to conduct DSC analysis. The synthesized samples were heated to 300 °C and kept fixed for 3–5 min to eliminate their history and then cooled as fast as possible to have an amorphous substance using a heating rate of 10 °C/min to evaluate Tg, Tm, cold crystallization, and thermal behavior. On the other hand, to analyze the melt (hot) crystallization, the samples were cooled with the specified cooling rate (Ф) after 5 min at 300 °C, and the quantities of heat flow and temperature were recorded. The thermal gravimetry analysis (TGA) of samples was performed using the STARe SW 9.10 Mettler Toledo instrument. Heating rate of samples was 10 °C/min.
Results and discussion
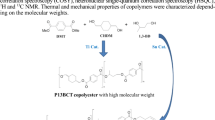
All samples were prepared based on the procedure explained in the experimental section. Table 2 gives the measured values regarding the characteristics of the synthesized samples. The samples are polyethylene terephthalate- co-ethylene [(6-Oxido-6H-dibenz[c, e][1,2]oxaphosphorin-6-yl) methyl] butanedioate (PET-co-PEDDP). For the sake of simplicity, the samples coded as PET-co-DDPnm, in which nm demonstrates the weight percent of phosphorus element in the polymer.
Number-average molecular weight was approximated using the intrinsic viscosity based on the following equation [27]:
All samples have enough high IV, molecular weight, and melting point, which are the indications of polymer formation. Based on the data reported in Table 2, all samples have AN less than 35 mg KOH/g, DEG less than 3 wt%, and COOH less than 25, which means acceptable polyester samples produced from the commercial point of view.
FTIR spectra
Figure 2 represents the FTIR spectra of samples with different phosphorus contents. The presented spectra report all peaks, which their assignment is given in Table 3 [28]. The detected bonds reported in Table 3 corroborated the synthesis of the polyethylene terephthalate-based copolyesters.

FTIR spectra of all prepared samples at different contents of phosphorus element
As well known, DDP has two fingerprint peaks around 760 and 1615 cm−1 that belong to the vibration of carbon–carbon double bond (C=C) [29] shown in Fig. 3. As can be seen, the size of the peak is growing by adding more DDP. Therefore, higher DDP amounts are incorporated in the chain with increasing DDP content.

Magnified images of FTIR peaks associated with DDP content
HNMR analysis
Figure 4 manifests the chemical structure of PET-co-DDP copolyester with illustrating the corresponding protons. The shifts related to this copolyester are reported in Table 4 [20]. Figure 5 also depicts the HNMR spectrum of PET-co-DDP07 copolyester.

Chemical structure of PET-co-DDP copolyester with corresponding protons

HNMR Spectrum of PET-co-DDP07 copolymer
From the stochiometric view, each diacid requires one dialcohol to form polyester. Hence, the area of diacids peaks should be equal to the area of dialcohols. To prove this claim, the following mathematical calculations are done:
As can be observed, these two values are close enough. Moreover, the area of d and g protons should be equal, too:
It is possible to calculate the number-average molecular weight based on the following computations:
The comparison of this quantity with the given value in Table 2 suggests that the parameter of \(\overline{M}_{n}\) in Table 2 could be utilized qualitatively.
Thermal behavior
Table 5 gives results of thermal features of the samples heated from the glassy state (amorphous) and, consequently, can measure the cold crystallization. To calculate the relative crystallinity, the heat of fusion for 100% crystalline (\(\Delta H_{m}^{0}\)), e.g., 135.8 J/g, was presumed [30].
Figure 6 manifests the heating of all samples from the amorphous state (quenched samples). Each specimen presents a unique Tg that is related to the sample composition. In other words, there is no phase separation between ethylene terephthalate and ethylene [(6-oxido-6H-dibenz[c,e] [1,2] oxaphosphorin -6-yl)methyl] butanedioate (DDP) segments.

Heating thermograms from amorphous state
In Table 5, there is no sensible and strong change in Tg versus DDP weight fraction. Qualitatively speaking, the glass transition temperature reduces with an increase in the DDP content [31, 32].
However, dissimilar to Tg (even inverse of Tg), the cold crystallization temperature, Tcc, and the melting point, Tm, of all samples exhibited a linear change. Figure 7 shows the remarkable linear change with excellent regression coefficients for the melting temperature. As it was reported [32, 33] and expected, melting temperature decreases with comonomer content due to chain disorder.

Melting point of all samples at different DDP weight fractions
Baur proposed the following equation to get the melting point for a copolymer with an ideal random structure:
where Tm, Tm0, xPET, Hf and R are melting point and equilibrium melting point temperatures, dominant monomer mole fraction, fusion enthalpy, and the universal gas constant, respectively. Figure 8 depicts 1/Tm against ln xPET plot which indicates an excellent fitting. Using data fitting method, the calculated values of Tm0 and Hf are 251.29 °C and 7.868 kJ/mol, respectively.

Melting point of all synthesized samples calculated by Baur’s equation
Also, Fig. 9 illustrates a reduction in the degree of crystallinity with an enhancement in the DDP wt%. The cold crystallization temperature, Tcc, is a measure of the polymer chains ability to be able to place in crystal structure [32]. The cold crystallization temperature in Fig. 10 shows that higher DDP content in the copolyester results in less crystallization ability of the sample caused by an increase in chain irregularity. Hence, cold crystallization happens in higher temperature.

Degree of crystallinity versus DDP weight fraction

Cold crystallization temperature versus DDP weight fraction
Non-isothermal melt (hot) crystallization kinetics, Hay model
The non-isothermal crystallization from the melt state has got practical interest in polymer processing and final articles manufacturing. Hence, a deep study of non-isothermal crystallization kinetics was performed. Various models and approaches have been introduced to study the kinetics of crystallization of polymers with emphasis on polyesters [34]. Modified Avrami, Velisaris and Seferis and Hay are some of the suggested models in the literature. Specially, it is well known that polyesters have primary as well as secondary crystallization. Figure 11 compares crystallization trend of various samples from the melt state at cooling rate equal to 10 °C. This figure was plotted in 3-dimensional form to present changes in a glance. Most of changes are due to secondary crystallization of samples. Some samples show bimodal curves which are contributed to the secondary crystallization.

Comparison between the crystallization behavior of all synthesized samples at Ф = 10 °C
It is essential to calculate the relative crystallinity (\(\chi_{t}\)) to analyze the non-isothermal crystallization kinetics. This parameter can be computed using DSC results, based on the following equation:
where dH/dt is the heat flow rate, and Ф is the cooling rate.
Various models have been introduced for describing the non-isothermal kinetics of crystallization. The Avrami model has been applied widely. The detailed examination of kinetic study for various models is given in supplementary materials. Other models mostly try to improve the Avrami model. Moreover, it is known that polyesters go under the primary as well as secondary crystallization. Improvement, correction, presentation, and application of new models is an ongoing research activity. The Hay model was selected as the best mode to analyze the crystallization kinetics in the present study [35]:
where n and Zp are the composite power index and the rate constant for diffusion-controlled growth of the secondary process, respectively. Also, the parameters of Zp and ks are pre-coefficients. Inspection of Eq. 9 reveals that the Hay model consists of two terms. The first term is the Avrami model and the second term is the square root of time weighted of the first term which takes into account the secondary crystallization. As a result, the Hay model has the ability to contemplate the secondary crystallization. Due to the complex form of the model, the nonlinear regression was employed with suggesting a new approach based on the regression coefficient (R2).
Table 6 gives the parameters of the Hay model and the corresponding regression coefficient. Based on the obtained data, all fitted parameters lead to a very acceptable R2 value close to 1. Hence, it means that this model is good enough to contemplate both primary and secondary crystallization. In the literature, for the exponent, n, a wide range has been reported. The range of 2.5–6.5 suggests that thermal nucleation occurs in the primary crystallization step, producing a three-dimensional spherical structure growth. Most of the crystallization happens in this step. The change in n could indicate the differences in crystal growth geometry and the type of nucleation, since it is known that an n value of 3 indicates the spherulitic growth from instantaneous nuclei, whereas a value of 4 represents the spherulitic growth from sporadic nuclei. A higher value of n means more three-dimensional structures than two-dimensional ones [36]. The extensive change in n is a sign of the asymmetry in crystallization behavior; therefore, different rate parameters would be calculated that are not comparable directly. It is observed that for all samples exponent n starts from a large number in Ф = 5 °C/min that describes production of a 3-D structure. n decreases with cooling rate increase up to Ф = 40 °C/min. In the course of non-isothermal DSC, there are variations in nucleation and rate with respect to temperature which could be contributed to the secondary crystallization. The inspection of the curves shows that there is a roll-off of around 60% relative crystallinity. This roll-off is contributed to the secondary crystallization that causes the deviation from the Avrami plots, as shown in Fig. 12. Therefore, the Avrami model is not a good predictor for crystallization kinetics. In this way, nucleation seems to become time and cooling rate dependent. \(k_{{\text{s}}}\) is the square root of time coefficient. It is detected that for all samples with certain amount of DDP, \(k_{{\text{s}}}\) increases with cooling rate. Hence, it is concluded that secondary crystallization gets dominant with cooling rate increase. Some variations are realized among data. It could be contributed to the secondary crystallization and change in behavior that is observed in Fig. 11.

Double logarithmic curves of all synthesized samples predicted by Hay model
Figure 12 exhibits the double logarithm curves of all produced samples. The deviation of \(\log \left( { - \ln \left( {1 - \chi_{t} } \right)} \right)\) versus log t from the line is the indication of having the secondary crystallization. As shown, an increase in the DDP wt% up to 5% causes a deviation from the line, especially in lower cooling rates. However, there is less deviation from the line by increasing DDP wt% higher than 5%. Hence, The Hay model has ability to take into consideration the change in crystallization behavior and polymorphism.
The relative crystallization rate can be compared by the crystallization rate parameter (CRP) suggested by Zhang et al. [37]. The slope of t1/2 inverse versus Ф gives CRP that are reported in Table 7 for all samples and plotted in Fig. 13. A greater slope indicates a faster crystallization rate. Although there are some variations in CRP values among samples, there is no drastic change in it. In other words, effect of DDP incorporation in polymer chain is small on relative rate. Relative crystallinity plots, in supplementary material, reveal their similarities. Despite close CRP, \(t_{{{{1} \mathord{\left/ {\vphantom {{1} {2}}} \right. \kern-\nulldelimiterspace} {2}}}}\) increases with DDP content in all cooling rates. Therefore, it is concluded adding a big side group such as DDP to PET chain more or less increases non-isothermal crystallization half-time.

t1/2 inverse versus Ф for all synthesized samples
Ozawa model
Ozawa suggested the following equation to analyze the non-isothermal crystallization:
where \(\chi_{{\text{T}}}\) is the relative crystallinity at a specific temperature, K(T) relates to the overall crystallization rate, m is the Ozawa's exponent, associated with the crystal growth dimension. The parameters of this model could be calculated through the following logarithmic form:
Table 8 gives the calculated parameters of the Ozawa model for all studied samples. The details of the extra data are represented in the supplementary materials.
The inspection of regression coefficient shows that not all of them are acceptable, and in comparison with the Hay model, they are not that close to 1.0. With respect to Ozawa interpretation [38], his model is derived based on the thermoanalytical data and applied to small and close cooling rates for homopolymer, successfully. However, polymer goes under large cooling rates during the processing. Moreover, as it is observed in Fig. 12 secondary crystallization takes place for synthesized samples. Therefore, having the secondary crystallization causes the deviation from the Ozawa model.
Mo model
Mo et al. combined the Avrami and the Ozawa models to introduce the following kinetic equation [39]:
This model gives the cooling rate and crystallization time correlation. Table 9 lists the parameters of the Mo model calculated at different relative crystallinities of 0.2, 0.5, and 0.8.
Non-isothermal cold crystallization kinetics, Hay model
The cold crystallization analysis, which is the crystallization from the amorphous and glassy state, could furnish applicable information, particularly in heat treatment processes. For the sake of comparison, cold crystallization behavior was studied in one heating rate. Figure 14 presents the cold crystallization trends for all PET samples from the glassy state using a heating rate of 10 °C/min. It is interesting to consider that all thermograms exhibit a symmetric behavior. Therefore, primary crystallization is dominant in such a cold crystallization. Table 10 gives the results of the Hay fitting model for the cold crystallization. Moreover, Fig. 15 reveals the double logarithmic plots, which showing an approximately straight line for all samples.

Cold crystallization thermograms of all synthesized samples with Ф = 10 °C

Double plot for all synthesized sample using a heating rate of 10 °C/min
Thermogravimetric analysis
The thermogravimetric analysis is applied to investigate the DDP impact on the thermal stability of all samples [11]. Although TGA studies in more than one heating rate could be informative, it could be another research objective. Figure 16 depicts the TGA and corresponding DTG curves of neat PET and copolyesters. It distinguished that the TGA curves are very close to each other, so there is no meaningful difference. However, the utilization of the degradation kinetics could reveal the influence of incorporating DDP comonomer in the PET chain.

Thermogravimetric curves of neat PET and copolyesters
There are various methods to analyze TGA data and calculate the activation energy of thermal degradation. The fractional extent of the reaction is defined as:
where wi, wt, and wf are the initial mass, the mass at the time t, and the final mass, respectively. The general kinetics equation for the thermal degradation is the following:
where A,, and R are the pre-exponential factor, fractional extent of the reaction, and the universal gas constant, respectively. It is possible to rearrange the derivative of the fractional extent of the reaction:
Hence, by integrating the above equation:
Using the approximation in the Coats–Redfern method, the following equation is resulted [40]:
By plotting the left-hand side versus 1/T and fitting a line, the kinetics parameters were calculated and are given in Table 11. It should be emphasized that \(\left( {1 - {{2RT} \mathord{\left/ {\vphantom {{2RT} E}} \right. \kern-\nulldelimiterspace} E}} \right)\) is constant. Figure 17 illustrates a kind of these plots for neat PET. Therefore, Coats–Redfern method has ability to depict degradation kinetics.

Coats–Redfern equation plot for PET sample
Figure 18 gives the diagram of kinetics parameters. As can be detected, all kinetic parameters changed with DDP weight fraction.

Kinetics parameters of thermal degradation for all synthesized samples with different DDP contents
Conclusions
This research aims at fabricating PET and phosphorous-containing copolyesters through a two-step procedure; esterification; and polycondensation. Ethylene glycol (EG) and terephthalic acid (TPA) are the principal monomers, and [(6-oxido-6H-dibenz[c,e] [1,2] oxaphosphorin -6-yl)methyl]butanedioic acid (DDP) is a phosphorous-containing comonomer. The measurement of fundamental properties such as intrinsic viscosity and melting point verified the successful production of polymer. The structure of samples was studied by various analyses of FTIR, HNMR, and TGA. The DDP fingerprint peaks were observed in the FTIR spectra of all samples. The calculated peaks area for the PET-co-DDP07 specimen is consistent with its structure. Melting point (Tm) and degree of crystallinity are reduced approximately with a linear trend by increasing DDP content. The secondary crystallization appeared in the crystallization trends. The improved Avrami, Velisaris, and Hay models were also applied to describe the kinetics of crystallization. Based on the prediction accuracy, the Hay model could estimate the kinetics very well, even in the presence of secondary crystallization. The Ozawa and Mo models were employed for the non-isothermal crystallization. The Hay model has the excellent capability of describing both hot and cold crystallization. Also, the Coats–Redfern equation was utilized to examine the influence of comonomer on thermal degradation. The outcomes of the current research could apply to polymer processing to achieve a certain degree of crystallinity.
References
Scheirs J, Long TE (eds) (2005) Modern polyesters: chemistry and technology of polyesters and copolyesters. Wiley
Fakirov S (ed) (2002) Handbook of thermoplastic polyesters. Wiley
Ian Tiseo, Global production capacity of polyethylene terephthalate 2014–2024, Statista Research Department
Horrocks AR, Price D, Price D (eds) (2001) Fire retardant materials. Woodhead Publishing
Ibbotson C, Sheldon RP (1979) Heterogeneous crystallisation of polyethylene terephthalate. British Polym J (Polymer International) 11(3):146–150. https://doi.org/10.1002/pi.4980110308
Phang IY, Pramoda KP, Liu T, He C (2004) Crystallization and melting behavior of polyester/clay nanocomposites. Polym Int 53(9):1282–1289. https://doi.org/10.1002/pi.1513
Mohsen-Nia M, Memarzadeh MR (2013) Characterization and non-isothermal crystallization behavior of biodegradable poly(ethylene sebacate)/SiO2 nanocomposites. Polym Bull 70(8):2471–2491. https://doi.org/10.1007/s00289-013-0967-3
Kahkesh S, Rafizadeh M (2020) Flame retardancy and thermal properties of poly(butylene succinate)/nano-boehmite composites prepared via in situ polymerization. Polym Eng Sci 60(9):2262–2271. https://doi.org/10.1002/pen.25468
Agrawal H, Awasthi K, Saraswat VK (2014) Non-isothermal crystallization kinetics of TiO2 nanoparticle-filled poly(ethylene terephthalate) with structural and chemical properties. Polym Bull 71(6):1539–1555. https://doi.org/10.1007/s00289-014-1140-3
Lin X, Zhang H, Ke M, Xiao L, Zuo D, Qian Q, Chen Q (2014) Non-isothermal crystallization kinetics of poly(ethylene terephthalate)/mica composites. Polym Bull 71(9):2287–2301. https://doi.org/10.1007/s00289-014-1187-1
Gao W, Wang Z, Zhao Z, Ding L, Zhu Y (2017) Effect of barium sulfate on thermal stability and crystallization properties of poly (ethylene terephthalate). J Therm Anal Calorim 129(2):1047–1055. https://doi.org/10.1007/s10973-017-6237-0
Valapa R, Hussain S, Krishnan Iyer P, Pugazhenthi G, Katiyar V (2016) Non-isothermal crystallization kinetics of sucrose palmitate reinforced poly(lactic acid) bionanocomposites. Polym Bull 73(1):21–38. https://doi.org/10.1007/s00289-015-1468-3
Wang G, Chen Y (2018) Isothermal crystallization and spherulite morphology of poly(ethylene terephthalate)/Na+-MMT nanocomposites prepared through solid-state mechanochemical method. J Therm Anal Calorim 131(3):2611–2624. https://doi.org/10.1007/s10973-017-6857-4
Mohammadi S, Taremi FA, Rafizadeh M (2012) Crystallization conditions effect on molecular weight of solid-state polymerized poly(ethylene terephthalate). Iranian Polym J 21(7):415–422. https://doi.org/10.1007/s13726-012-0044-z
Papageorgiou G, Achilias D, Bikiaris D, Karayannidis G (2006) Isothermal and non-isothermal crystallization kinetics of branched and partially crosslinked PET: DSC study. J Therm Anal Calorim 84(1):85–89. https://doi.org/10.1007/s10973-005-7366-4
Saeed HA, Eltahir YA, Xia Y, Yimin W (2014) Non-isothermal crystallization kinetics and nucleation activity of hyperbranched polyester (HBPET) in recycled PET. Polym Bull 71(3):595–612. https://doi.org/10.1007/s00289-013-1080-3
Ravari F, Mashak A, Nekoomanesh M, Mobedi H (2013) Non-isothermal cold crystallization behavior and kinetics of poly(l-lactide): effect of l-lactide dimer. Polym Bull 70(9):2569–2586. https://doi.org/10.1007/s00289-013-0972-6
Wang LS, Wang XL, Yan GL (2000) Synthesis, characterisation and flame retardance behaviour of poly(ethylene terephthalate) copolymer containing triaryl phosphine oxide. Polym Degrad Stab 69(1):127–130. https://doi.org/10.1016/S0141-3910(00)00050-1
Wang CS, Shieh JY, Sun YM (1999) Phosphorus containing PET and PEN by direct esterification. Eur Polym J 35(8):1465–1472. https://doi.org/10.1016/S0014-3057(98)00234-1
Chang SJ, Chang FC (1999) Synthesis and characterization of copolyesters containing the phosphorus linking pendent groups. J Appl Polym Sci 72(1):109–122. https://doi.org/10.1002/(SICI)1097-4628(19990404)72:1%3c109::AID-APP12%3e3.0.CO;2-Q
Mohammadi Avarzman A, Rafizadeh M, Afshar Taromi F (2021) Branched polyester based on the polyethylene tere/iso phthalate and trimellitic anhydride as branching agent. Polym Bull 79(8):1–23. https://doi.org/10.1007/s00289-021-03802-x
Zhou C, Wei Z, Yu Y, Li Y (2015) Synthesis and crystallization behavior of novel poly(butylene succinate) copolyesters containing phosphorus pendent groups. J Therm Anal Calorim 120(3):1799–1810. https://doi.org/10.1007/s10973-015-4511-6
Naghavi Sheikholeslami S, Rafizadeh M, Afshar Taromi F, Shirali H (2017) Crystallization and photo-curing kinetics of biodegradable poly(butylene succinate-co-butylene fumarate) short-segmented block copolyester. Polym Int 66(2):289–299. https://doi.org/10.1002/pi.5264
Al-Mulla A (2012) Determination of crystallization parameters and occurrence of trans-reaction in a ternary polymer blend system. Polym Bull 69(9):1053–1071. https://doi.org/10.1007/s00289-012-0779-x
Kint DP, Muñoz-Guerra S (2003) Modification of the thermal properties and crystallization behaviour of poly(ethylene terephthalate) by copolymerization. Polym Int 52:321–336. https://doi.org/10.1002/pi.1175
Chen M, Wang HC, Ko CY, Chen RY, Wang CL, Tseng IM (2008) Crystallization kinetics and melting behavior of poly[(trimethylene terephthalate)-co-(38 mol% ethylene terephthalate)] copolymer. Polym Int 57(2):297–305. https://doi.org/10.1002/pi.2346
Sánchez-Arrieta N, De Ilarduya AM, Alla A, Muñoz-Guerra S (2005) Poly(ethylene terephthalate) copolymers containing 1, 4-cyclohexane dicarboxylate units. Eur Polym J 41(7):1493–1501. https://doi.org/10.1016/j.eurpolymj.2005.02.004
Charles J, Ramkumaar GR (2009) FTIR and thermal studies on polyethylene terephthalate and acrylonitrile butadiene styrene. Asian J Chem 21(6):4389
Zhang C, Wang J, Song S (2019) Preparation of a novel type of flame retardant diatomite and its application in silicone rubber composites. Adv Powder Technol 30(8):1567–1575. https://doi.org/10.1016/j.apt.2019.05.002
Jog JP (1995) Crystallization of polyethyleneterephthalate. J Macromol Sci Part C Polym Rev 35(3):531–553. https://doi.org/10.1080/15321799508014598
Wood LA (1958) Glass transition temperatures of copolymers. J Polym Sci 28(117):319–330. https://doi.org/10.1002/pol.1958.1202811707
Balachandar M, Balakrishnan T, Kothandaraman H (1983) Glass transition and melting temperatures of random copolyesters of poly (ethylene terephthalate) with p-hydroxybenzoic acid, 3. Die Makromolekulare Chemie Macromol Chem Phys 184(2):443–453. https://doi.org/10.1002/macp.1983.021840218
Wang ZG, Hsiao BS, Sauer BB, Kampert WG (1999) The nature of secondary crystallization in poly(ethylene terephthalate). Polymer 40(16):4615–4627. https://doi.org/10.1016/S0032-3861(99)00067-1
Kelly CA, Hay JN, Turner RP, Jenkins MJ (2020) The effect of a secondary process on the analysis of isothermal crystallisation kinetics by differential scanning calorimetry. Polymers 12(1):19. https://doi.org/10.3390/polym12010019
Chen Z, Hay JN, Jenkins MJ (2016) The effect of secondary crystallization on crystallization kinetics–polyethylene terephthalate revisited. Eur Polym J 81:216–223. https://doi.org/10.1016/j.eurpolymj.2016.05.028
Gaonkar AA, Murudkar VV, Deshpande VD (2020) Comparison of crystallization kinetics of polyethylene terephthalate (PET) and reorganized PET. Thermochim Acta 683:178472
Zhang R, Zheng H, Lou X, Ma D (1994) Crystallization characteristics of polypropylene and low ethylene content polypropylene copolymer with and without nucleating agents. J Appl Polym Sci 51(1):51–56
Ozawa T (1971) Kinetics of non-isothermal crystallization. Polymer 12(3):150–158
Liu T, Mo Z, Wang S, Zhang H (1997) Nonisothermal melt and cold crystallization kinetics of poly(aryl ether ether ketone ketone). Polym Eng Sci 37(3):568–575. https://doi.org/10.1002/pen.11700
Regnier N, Guibe C (1997) Methodology for multistage degradation of polyimide polymer. Polym Degrad Stab 55(2):165–172. https://doi.org/10.1016/S0141-3910(96)00115-2
Funding
No funding was received for conducting this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Moghadam, A.S.N., Rafizadeh, M. & Taromi, F.A. Preparation of poly(ethylene terephthalate) copolyester with phosphorus-containing comonomer: characterization, thermal behavior, and non-isothermal crystallization kinetics. Polym. Bull. 80, 8151–8180 (2023). https://doi.org/10.1007/s00289-022-04447-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-022-04447-0