
Abstract
A kind of polyurethane rubber (PU)/n-octadecane (n-OD)@silicon dioxide (SiO2)-polymethyl acrylate (PHEMA) form-stable phase change material (PCM) was fabricated in this paper. n-OD@SiO2-PHEMA microcapsules were prepared in one-pot, through interfacial hydrolysis polycondensation of alkoxy silanes and radical polymerization of HEMA. Then, n-OD@SiO2-PHEMA microcapsules were added into PU matrix to obtain PU/n-OD@SiO2-PHEMA composites. SiO2-PHEMA hybrid shell not only provides a shelter for core material, but also increases interfacial bonding between microcapsules with PU and effectively improves the mechanical properties of composites. The morphologies, chemical compositions and crystal structures of microcapsules and composites were characterized by SEM, TEM, FT-IR and XRD methods. The thermal and mechanical properties of the microcapsules and composites were characterized by differential scanning calorimetry and mechanical performance tests. The results show that the thermal and mechanical properties of PU/n-OD@SiO2-PHEMA composites were better than those of neat PU. The PU/n-OD@SiO2-PHEMA composites endow the microcapsules with improved thermal reliability and leakage proof property. The composites were found as form-stable PCM with low leakage rate. The phase change latent heat of the PU/n-OD@SiO2-PHEMA composites was up to 85.1 J/g and the composites can be used for thermal energy storage.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Growing of world population and intense industrial consuming have created the shortage of energy. Meanwhile, toxic gases produced by fossil fuels have caused global warming and the collapse of ecosystems. Thus, the development of green and sustainable energy science should be our urgent mission. In recent years, investigation of new energy has drawn the extensive consideration of researchers. For example, solar energy is a kind of abundant resource and photocatalysis is a novel and eco-friendly technology [1,2,3,4]. Converting solar energy to chemical energy in the form of hydrogen through nanoparticles such as TiO2 [5] and Fe2O3 [6] is regarded as a promising approach to solve the environment problem such as fuel pollution in the textile industry [7, 8]. In addition, thermal energy storage (TES) [9] possessing high-efficiency energy storage methods and recovery efficiency has drawn the attention of researchers. Phase change materials (PCMs) are mainly divided into three transformation modes: solid–gas, solid–liquid and liquid–gas. Solid–gas and liquid–gas will disclose gas during energy conversion, which is harmful to energy storage and conversion. The solid–liquid transition is a relatively mature option [10]. However, high leakage rate during the phase transition restricts their applications. Encapsulating solid–liquid PCMs into micro/nano-sized tiny containers can effectively improve the leakage problem, and it can also improve heat transfer by increasing the surface area to volume ratio [11, 12]. Researchers use different shell materials to encapsulate solid–liquid PCMs, organic shells such as melamine–formaldehyde resin (MF) [13, 14], polymethyl methacrylate (PMMA) [15,16,17], polyurea resin (Polyurea) [18] and polystyrene (PS) [19]. Inorganic shells such as calcium carbonate (CaCO3) [20] and silicon dioxide (SiO2) [21,22,23]. Zhu et al. [24] used silicon dioxide to microencapsulate n-octadecane to prepare PCMs and used styrene and hydroxyethyl methacrylate to prepare a polymer-SiO2 hybrid shell. The polymer shell had good thermal stability and tightness, which improved the leakage of n-octadecane at phase transition temperature.
In addition, the form-stable PCM [25] can also effectively solve the leakage problem of solid–liquid PCM, such as inorganic support material diatomaceous earth [26] and organic support material polyurethane [27]. The combination of microcapsules and support materials can have the advantages of the above two methods. For example, Guo [28] successfully used the interfacial hydrolysis polycondensation reaction of tetraethylorthosilicate and γ-aminopropyltriethoxysilane in the miniemulsion to coat paraffin wax and used the prepared microcapsules to fill silicone rubber to obtain the form-stable silicone rubber/paraffin@silicon. The form-stable PCMs filled with microcapsules effectively enhanced the mechanical properties of silicone rubber and possessed high enthalpies, good thermal cycling and great applications in thermal energy storage. To the best of our knowledge, however, many works hardly report to prepare the form-stable PCM with organic–inorganic hybrid shell microcapsules. Compared with the single shell microcapsules, organic–inorganic hybrid shell microcapsules have advantages in thermal conductivity and compatibility with support materials.
Here, we present a kind of PU/n-OD@SiO2-PHEMA form-stable phase change material. According to the literature, PU is a kind of rubber with high density, high hardness, good elasticity and good biocompatibility. However, the weak interaction force between macromolecules caused poor mechanical properties that limited its application. Inorganic fillers and organic polymer are chosen to modify PU, such as SiO2 [29], CaCO3 [30], acrylic resin [31] and epoxy resin [32]. SiO2 has a huge specific surface area, which can provide strong van der Waals force between the filler and the polymer chain, while acrylic resin can effectively improve the thermal stability. In this study, the sulfhydryl-modified n-OD@SiO2 microcapsules were first prepared by the miniemulsion method, and then, the sulfhydryl groups on the surface of SiO2 and the double bonds in hydroxyethyl methacrylate were used for free radical polymerization to obtain n-OD@SiO2-PHEMA hybrid shell microcapsules. Finally, the microcapsules were added to the PU matrix to prepare PU/n-OD@SiO2-PHEMA composites. In this composite, n-OD makes PU/n-OD@SiO2-PHEMA composites possessing latent heat of phase change, PU and SiO2-PHEMA hybrid shell provide dual encapsulation for n-OD. The prepared PU-based form-stable PCMs have thermal energy storage capacity, low leakage rate and good mechanical properties.
Experimental methods
Materials
N-octadecane (n-OD, 98 wt%), γ-mercaptopropyltriethoxysilane (KH-590) and hydroxyethyl methacrylate (HEMA) were purchased from Aladdin. Tetraethyl orthosilicate (TEOS), anhydrous ethanol (NH3·H2O), 2,2′azobisiso butyronitrile (AIBN), trimethylolpropane (TMP) and cetyltrimethylammonium bromide (CTAB) were purchased from Sinopharm Chemical Reagents. HEMA were purified by passing through a neutral alumina column, to remove retardants. AIBN was recrystallized from 95% ethanol before use. Two dibutyltin dilaurate (DBT) was purchased from Beijing Zhengheng Chemical Co., Ltd. Diisocyanated’isophorone (IPDI) was commercially supplied by Jinan Huakai Resin Co., Ltd. Poly (1,4-Bytylene adipate) (Mn = 1000) was obtained from Shandong Jiaying Chemical Technology Co., Ltd. All chemicals were of reagent quality and used without further purification, unless stated otherwise.
Fabrication of n-OD@SiO2-PHEMA hybrid shell microcapsules
Typically, n-OD (10.0 g), TEOS (8 mL), KH-590 (3 mL) and purified AIBN (0.1 g) were mixed in a breaker (500 mL), then deionized H2O (145 mL), absolute ethanol (72.5 mL), CTAB (1.65 g) and HEMA (5 mL) were added into the breaker and heating at 30 °C to form a clear solution. The solution was emulsified by means of FJ200-S homogenizer at 10,000 r/min for 5 min and ultrasounded for 10 min to form a stable miniemulsion; then, the solution was poured into a three-necked flask (500 mL) equipped with a mechanical stirrer. The reaction was catalyzed by 30 mL dilute ammonia solution and stirred at 35 °C for 2 h at a rate of 330 rpm. Then, the temperature was raised to 80 °C. The radical polymerization was initiated by AIBN and continued for 4 h. Finally, the white powder was obtained after filtration, washing and freeze drying. The fabrication procedure is shown in Scheme 1. The surface of n-OD droplets are full of CTAB micelles [33], which provides a layer of positive charges. The positive charges have a charge attraction on the negatively charge SiO2, which pledges SiO2 to be deposited on the surface of the n-OD droplets to form n-OD@SiO2 microcapsules. The modified SiO2 was obtained through hydrolyzation and polycondensation of silanol (KH-590) and TEOS. Finally, n-OD@SiO2-PHEMA hybrid shell microcapsules were obtained through the free radical polymerization mechanism between -SH and double bond.

Fabrication process of n-OD@SiO2 and n-OD@SiO2-PHEMA
Preparation of PU/n-OD@SiO2-PHEMA composites
The n-OD@SiO2-PHEMA microcapsules as a kind of filler with phase change materials were added into the PU matrix. TMP acted as a crosslinking agent and DBT used as a catalyst, the fabrication procedure is shown in Scheme 2. In this procedure, a certain amount of PBAG (Mn = 1000) and redundant IPDI reacted for 2 h to prepare -NCO group capped PU prepolymer. Then n-OD@SiO2-PHEMA microcapsules were gradually added into the PU prepolymer. TMP and DBT were added into the mixture after the microcapsules well stirring with PU. Subsequently, the mixture was poured into a Teflon mold to solidify for 24 at room temperature to acquire composites. The -OH (PHEMA and SiO2) on the surface of microcapsules could form chemical crosslinks with the PU prepolymer to increase the bonding between microencapsules and PU matrix. Composites with different n-OD@SiO2-PHEMA contents were prepared and labeled as PU/n-OD@SiO2-PHEMA-40, 60, 80, 100, respectively. Meanwhile, PU, PU/n-OD@SiO2 were prepared for comparison.

Fabrication process of PU/n-OD@SiO2-PHEMA composites
Characterization
Fourier transform infrared (FT-IR)
The chemical composition of microcapsules and composites was analyzed by Nicolet 560 FTIR spectrometer (iS10 Thermo Nicolet Co., Ltd, Madison-n, America) with the KBr sampling method and transmission method, respectively. The range of the spectra was from 4000 to 400 cm−1.
X-ray powder diffraction (XRD)
The crystal diffraction peak of microcapsules and composites were confirmed by high-resolution X-ray diffractometer (D/MAX2500, Japan science smartlab 9). The scanning speed was 4°/min at 2θ range of 5–80°, and the step was 0.02.
Particle size analyzer
The diameters of the prepared n-OD@SiO2 and n-OD@SiO2-PHEMA microcapsules were measured by Malvern Mastersizer 3000 (UK). The microencapsules (0.05 g) were dispersed in absolutely ethanol (10 mL), and the results were obtained by taking the average of three measurements.
Field emission scanning electron microscope (FE-SEM)
The characterization of the microcapsules and composites was conducted on a SUPRA55 field emission scanning electron microscope. The cross section of the composites sheet was obtained by brittle fracture in liquid nitrogen, which was then installed on the sample table and sprayed with gold before testing. During the SEM test, the cross sections of all samples were directly below the electron beam (install the samples vertically to view the cross section).
Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) was performed on a JEOL 2100 transmission electron microscope operated at an accelerating voltage of 100 kV. The microcapsules were dispersed absolutely ethanol, and a drop of the mixture was dripped to copper mesh (100 mesh). The thickness of shell was determined based on the obtained TEM micrographs.
Differential scanning calorimetry (DSC)
The thermal properties of n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA and PU/n-OD@SiO2-PHEMA composites were performed by a DSC-204-F1 (GE) differential scanning calorimetry (DSC), the heating or cooling rate was 10 °C/min and the temperature range was from 0 to 60 °C in N2 atmosphere, and the weight of sample was about 5 mg.
Dynamical mechanical analysis (DMA)
The glass transition of composites was performed on a DMA 8000 (Llantrisant. CF72 8YW. UK). The method of test is tensile mode, the length, width and thickness of each example was 10 mm, 5 mm and 0.5 mm. Composites were measured at 5 °C/min rate from − 10 °C to 120 °C.
The thermogravimetric analysis (TGA)
The thermogravimetric analysis (TGA) was performed on a TG-209-F-1 (USA) thermal analyzer and measured at 20 °C/min rate from 35 to 600 °C. The analyses were taken in an inert nitrogen atmosphere at a 100 mL/min flow rate. Each sample was about 3 mg.
Leakage test
The stability of the microcapsules and the form-stable PCMs was evaluated by measuring the leakage rate (Lr%). In this test, the samples of initial weight referred to Minitial were placed on filter papers to observe the leaked n-OD when they were heated to 60 °C (60 °C, which is higher than the melting temperature of n-OD) in the Vacuum oven. Keeping to replace another filter paper until the weight of samples no longer changed. The final remaining weight of the samples were labeled as Mremainder. The leakage rate of the samples was calculated by the following equation:
Mechanical performance test
The tensile strength and elongation at break of the dumbbell-shaped samples were measured by the MTS industrial system C45.504 universal testing machine. Dumbbell specimens was obtained from Teflon molds. The effective thickness and length of the dumbbell specimens were 2 mm and 15 mm, respectively. At least five measurements were taken for each sample at room temperature (RT), and all experiments were performed at 100 mm/min. By using a shore hardness tester to test the hardness of the sample. Five results were taken for each sample at room temperature and 60 °C. All data are the average of five measurement results for each sample.
Results and discussion
Characterization of n-OD@SiO2 and n-OD@SiO2-PHEMA microcapsules
See Fig. 1.

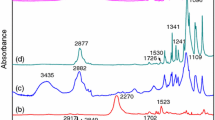
a FT-IR spectra of n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA; b XRD patterns of n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA
The topography and particle size analysis of microcapsules.
The chemical compositions of the microcapsules were confirmed by FT-IR characterization, as shown in Fig. 1a. The FR-IR spectra of the microcapsules (n-OD@SiO2, n-OD@SiO2-PHEMA) show the similar characteristic peaks of core material n-OD (2923 cm−1, 2853 cm−1, 1469 cm−1, 1370 cm−1, 718 cm−1). The microcapsules also have the peaks at 1086 cm−1 and 467 cm−1, which are the characteristic absorption peaks of SiO2. The peak at 1731 cm−1 corresponds to stretching vibrational peak of C=O, indicating that the polymer (PHEMA) was successfully grafted on the surface of SiO2. And the characteristic absorption peak at 3436 cm−1 corresponds to -OH is shown in n-OD@SiO2 and n-OD@SiO2-PHEMA, which is owing to the -OH groups from SiO2 and PHEMA. Therefore, the formation of n-OD@SiO2-PHEMA was clearly demonstrated. In addition, by comparing the XRD patterns of n-OD and microcapsules in Fig. 1b, it can also be shown that the core material n-octadecane was encapsuled in the microcapsules and displayed a triclinic phase structure similar to n-octadecane.
The morphology of n-OD@SiO2 and n-OD@SiO2-PHEMA microcapsules is shown in Fig. 2. n-OD@SiO2 are obviously regular spherical (Fig. 2a), and as can be seen from the TEM (Fig. 2d) picture, the shell thickness of the n-OD@SiO2 microcapsule is about 25 nm, indicating that SiO2 encapsuled the core material n-octadecane greatly, and the particle size is 326 nm (Fig. 2g). But the microcapsules have some certain folds (Fig. 2d) due to the evaporation of solvent in the microcapsules, demonstrates that the SiO2 is mesoporous. Compared with the SiO2 microcapsules, the polymer-SiO2 shell reacted with the mesoporous SiO2 wall (Fig. 2b, c), the n-OD@SiO2-PHEMA is dense and smooth, it owns a thicker shell (Fig. 2e, f, 170 nm) and larger particle size (Fig. 2h, 1.68 μm). The C, Si, O and S elements are clearly seen from the EDS analysis of microcapsules (Fig. 2i), which further demonstrate that n-OD@SiO2-PHEMA have been successful. But the TEM image of the n-OD@SiO2-PHEMA seems not to be perfectly spherical, this problem could be solved if we consider the contents of PHEMA to regulate the shape of the microcapsules, we will have the chance to discuss the situation in future experiments. Therefore, despite the SEM and EDS preliminary characters, they can clearly indicate n-OD@SiO2 and n-OD@SiO2-PHEMA have been prepared successfully.

SEM images of a n-OD@SiO2 and b, c n-OD@SiO2-PHEMA; TEM images of d n-OD@SiO2 and e, f n-OD@SiO2-PHEMA; Particle size distribution of g n-OD@SiO2 and h n-OD@SiO2-PHEMA. i EDS images of n-OD@SiO2 and n-OD@SiO2-PHEMA
Chemical composition and Crystallization performance of composites
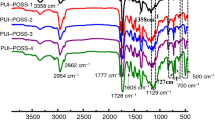
The structures and chemical compositions of the prepared form-stable PCMs are analyzed by FT-IR and XRD methods. As shown in Fig. 3a FT-IR spectrum, the characteristic absorption peaks of neat PU and n-OD@SiO2-PHEMA microcapsules can be clearly observed in the curve of PU/n-OD@SiO2-PHEMA composites. The N–H group (3300 cm−1) in PU and the -OH group (3438 cm−1) in n-OD@SiO2-PHEMA microcapsules form hydrogen bond crosslinks, hydrogen bonding in PU composites is also of great interest since it plays an important role in determining phase segregation [34]. Its hard segment increased with the addition of filler, and the degree of hydrogen bonding also increased. The peak intensity of –OH groups (3438 cm−1) are weaken in the composites and then redshifted (3366 cm−1). The strong absorption peak at 1730 cm−1 corresponds to the C=O group in PU. In addition, PU/PHEMA/n-OD@SiO2 composites show the same characteristic absorption peaks as the core material n-octadecane (2923 cm−1, 2853 cm−1, 1466 cm−1, 1390 cm−1 and 719 cm−1), and the characteristic absorption peaks of Si–O–Si appear at 1030 cm−1 and 460 cm−1, which prove that the PU matrix material had successfully encapsuled n-OD@SiO2-PHEMA microcapsules.

a the FTIR spectra of n-OD, n-OD@SiO2-PHEMA, PU/n-OD@SiO2-PHEMA and PU; b XRD patterns of PU/n-OD@SiO2-PHEMA-40, 60, 80, 100 and PU
The crystal performance of neat PU, n-OD@SiO2-PHEMA, PU/n-OD@SiO2-PHEMA composites are presented in Fig. 5b. PU shows amorphous peak and manifests its structureless. In addition, the shape of crystal peak in all PU/n-OD@SiO2-PHEMA composites is similar with n-OD@SiO2-PHEMA microcapsules, which indicates n-OD@SiO2-PHEMA was successfully encapsulated in these composites and the composites possess the same triclinic crystal structure compared to n-OD.
SEM topography of PU and composites
The cross-sectional SEM pictures of neat PU, PU/n-OD@SiO2, PU/n-OD@SiO2-PHEMA-40 and PU/n-OD@SiO2-PHEMA-100 are shown in Fig. 4. It is obvious that the cross section of PU is smooth (Fig. 4a). However, PU/n-OD@SiO2 is clearly visible that n-OD@SiO2 was partly reunited (Fig. 4b), it is demonstrated that n-OD@SiO2 badly dispersed in the PU matrix because of the poor compatibility between the n-OD@SiO2 and PU matrix. As can be seen from Fig. 4c, d, many hemispherical particles are observed on the fracture surface of PU/n-OD@SiO2-PHEMA, proving the existence of the n-OD@SiO2-PHEMA microcapsules. And it is consistent with the formula that the density of the microcapsules in the PU/n-OD@SiO2-PHEMA-100 composite is higher than that of the PU/n-OD@SiO2-PHEMA-40 composite. In short, it can effectively prevent n-OD from leaking at the working environment temperature through the dual encapsulation of PU matrix and microcapsules.

Cross-sectional SEM images of a neat PU, b n-OD@SiO2-PHEMA, c PU/n-OD@SiO2-PHEMA-40, d PU/n-OD@SiO2-PHEMA-100
Phase change properties
The thermal energy storage/release performance of the n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA and the PU/n-OD@SiO2-PHEMA composites are characterized by DSC, and the DSC curves of melting and crystalling process are shown in Fig. 5. For the two types of microcapsules (Fig. 5a), one-step melting occurred at 31.6 °C and 33.4 °C, respectively, which is lower than n-OD (34.3 °C). The reason is that whatever SiO2-shell and SiO2-PHEMA hybrid shell both provide a barrier for n-OD to limited heat conduction. It is worth noting that supercooling of microcapsules is lower than that of n-OD, explaining that the encapsulation of n-OD effectively delays the rate of cooling and enhances crystal perfection. As can be seen in Fig. 5b, it is consistent for curves of the composites, and with the content of n-OD@SiO2-PHEMA increasing on PU matrix, the enthalpies of the composites increase. The main statistics are shown in Table 1, the encapsulated efficiency of n-OD@SiO2-PHEMA (56.7%) has almost no changes from that of n-OD@SiO2 (56.8%) when was attributed to PHEMA, but the enthalpy of n-OD@SiO2-PHEMA (145.7 J/g) is higher than that of n-OD@SiO2 (141.9 J/g), indicating the thermal conductivity of n-OD@SiO2-PHEMA superior to n-OD@SiO2. The melting enthalpy of PU/n-OD@SiO2-PHEMA-40, 60, 80, 100 composites is up to 42.6 J/g, 58.1 J/g, 72.6 J/g, 85.1 J/g. For PU/n-OD@SiO2-100 composite, the content of n-octadecane on PU matrix had reached to 33.7% according to calculation, it provides the function of thermal energy storage for the form-stable PCMs greatly.

DSC curves of a n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA, b PU/n-OD@SiO2-PHEMA-40, 60, 80, 100 composites
Dynamic thermodynamic analysis
Dynamic mechanical properties of composites were characterized by DMA 8000, and the results are shown in Fig. 6. The glass transition temperature of neat PU is around 40 °C (Fig. 6a), and the glass transition of composites slightly decreased 5 °C. On the one hand, the free volume of composites is increased after filler, which results in the decrease of Tg. On the other hand, the filler can increase the degree of crosslinking in the composites, which block the movement of the macromolecular chains. However, the storage modulus and loss modulus (Fig. 6c, d) of composites are both higher than those of neat PU during the whole test temperature range due to the filler, indicating that fillers can improve the dynamic mechanical properties of PU composites in the highly elastic region.

DMA curves of a neat PU, b PU/n-OD@SiO2-40、PU/n-OD@SiO2-PHEMA-40、PU/n-OD@SiO2-PHEMA-80、PU/n-OD@SiO2-PHEMA-100; c storage modulus curves and d loss modulus curves
Thermal reliability
As shown in Fig. 7, the thermal stability of n-OD, n-OD@SiO2, n-OD@SiO2-PHEMA and PU/n-OD@SiO2-PHEMA composites is assessed by the TG method, and the statistics are listed in Table 2. Compared with the TG data of n-octadecane in Fig. 7a, n-OD@SiO2 microcapsules also represent a one-step thermal decomposition behavior in the curve of TG and represent a higher thermal decomposition temperature than pure n-octadecane because of the surface of n-octadecane filled with a layer of tight SiO2 shell. The weight loss of n-OD@SiO2-PHEMA microcapsules is divided into two steps. The first step (100–250 °C) is mainly due to the decomposition of the core material (n-OD), and the second step (300–500 °C) is the decomposition of the polymer shell (PHEMA). Two-step thermal decomposition is also observed in PU/n-OD@SiO2-PHEMA composites in Fig. 7b. Similarly, the first step is attributed to the core material (n-OD, 100–250 °C), but the second step is the decomposition of PU (300–500 °C), and the second decomposition temperature of PU/n-OD@SiO2-PHEMA composites is higher than that of neat PU, demonstrating that the SiO2-PHEMA hybrid shell effectively improves the stability of PU matrix. Because hydrogen bonding between filler and hard segments of PU is usually considered the strongest secondary force [35] that can display microphase separation, thereby exhibiting excellent thermal stability. Worthing observed, with the amounts of n-OD@SiO2-PHEMA microcapsules increasing in the PU matrix, the proportion of the first-step decomposition also increases, and the content of first-step decomposition in PU/n-OD@SiO2-PHEMA composites is consistent with the results of SEM confirmed.

TG curves of a OD, n-OD@SiO2, n-OD@SiO2-PHEMA; b PU/n-OD@SiO2-PHEMA-40, 60, 80, 100
Thermal conductivity
In order to examine the thermal cycle reliability of the microcapsules and PU/n-OD@SiO2-PHEMA composites, three samples (n-OD@SiO2, n-OD@SiO2-PHEMA, PU/n-OD@SiO2-PHEMA-100) were, respectively, heated to 60 °C and kept for 3 min, then cooled to 0 °C and kept for 3 min. After 10, 20, 30 heating–cooling cycles, the thermal reliability performance was characterized by DSC. It is found that the melting and crystallizing temperatures of microcapsules and PU/n-OD@SiO2-PHEMA have a good coincidence from the 10th cycles to the 20th and 30th cycles (as shown in Fig. 8a–c). Compared with the data of n-OD@SiO2-PHEMA, n-OD@SiO2 and PU/n-OD@SiO2-PHEMA-100 both appear three crystalling peaks, which are based on heterogeneous nucleation and homogeneous nucleation. The SiO2-PHEMA hybrid shell provides n-OD from revealing at operating temperature and improves undercooling. However, as n-OD@SiO2-PHEMA added to the PU prepolymer, PHEMA could react with the excess -NCO groups, explaining the reason why PU/n-OD@SiO2-PHEMA-100 appeared three crystalling peaks. Whatever melting enthalpies and crystalling enthalpies (Fig. 8d, e), PU/n-OD@SiO2-PHEMA-100 did not change much and possessed better stable thermal cycle performance than n-OD@SiO2 and n-OD@SiO2-PHEMA, indicating that composites exhibit excellent thermal reliability.

DSC curves of a n-OD@SiO2, b n-OD@SiO2-PHEMA, c PU/n-OD@SiO2-PHEMA-100 after experiencing 10, 20 and 30 heating–cooling cycles, d the melting enthalpies curves of samples, e the crystallizing enthalpies curves of samples
Mechanical properties
The mechanical properties of PU, PU/n-OD@SiO2, PU/n-OD@SiO2-PHEMA-40, 60, 80, 100 were tested (tensile strength, elongation at break and hardness test), and data are presented in Table 3. As shown in Fig. 9, the tensile strength and elongation at break of PU/n-OD@SiO2 are 11 Mpa and 526%, respectively, which are higher than those of PU (tensile strength, 9.72 MPa, elongation at break, 326%), due to the presence of n-OD@SiO2 playing a positive effect on PU. Meanwhile, the tensile strength and elongation at break of PU/n-OD@SiO2-PHEMA-40 reach 11.74 MPa and 530%, indicating that n-OD@SiO2-PHEMA microcapsules as fillers can effectively improve the performance of PU. And as the content of microcapsules increases, the mechanical properties of PU/n-OD@SiO2-PHEMA composites are enhanced. The tensile strength of PU/n-OD@SiO2-PHEMA-100 composites is up to 12.98 MPa, higher than that of PU. The hydrogen bonding interaction between filler and PU improves the degree of microphase separation, thereby improving composites mechanical properties [36]. The hardness can show the compressibility of materials. From the hardness data in Table 3, it is obviously that the room temperature hardness of PU/n-OD@SiO2 and PU/n-OD@SiO2-PHEMA composites is increased compared to neat PU. The hardness of the samples treated at 60 °C is lower than that of the samples at room temperature. Solid n-OD turns into the liquid caused to the decrease of the hardness when the samples heated above the phase transition temperature, which is critical to the heat dissipation process of the phase change materials in working environment.

Mechanical properties of pure PU, PU/n-OD@SiO2 and PU/n-OD@SiO2-PHEMA-40, 60, 80, 100
Leaking rates
The leakage rate properties of the microcapsules and form-stable PCMs (PU/n-OD@SiO2-PHEMA-40, 60, 80,100) were tested by treating them at 60 °C in an oven for 500 h. The weight loss of the samples was plotted against the heating time in Fig. 10a. It is shown that the weight loss is rapid in the initial 100 h due to the evaporation of solvent, and then, it turns slow while the heating time has reached 100 h. The n-OD@SiO2 microcapsules performed faster weight loss than n-OD@SiO2-PHEMA microcapsules, which proved that the hybrid shell provided a stronger barrier for n-OD. For the PU/n-OD@SiO2-PHEMA composites, with the content of microcapsules increasing in the PU matrix, the leakage rate was also increasing, and their melting enthalpies before and after heated for 500 h were measured and illustrated in Fig. 10b. The drop percentage of melting enthalpy follows such an order: PU/n-OD@SiO2-PHEMA-40 < PU/n-OD@SiO2-PHEMA-60 < PU/n-OD@SiO2-PHEMA-80 < PU/n-OD@SiO2-PHEMA-100 < n-OD@SiO2-PHEMA < n-OD@SiO2. Obviously, the order of leakage proof property is just inverse to the above-mentioned order. Therefore, it can be concluded that the PU matrix and polymer-SiO2 shell possess better leak-proof propertity.

a Leakage rate curves of microcapsules and PU/n-OD@SiO2-PHEMA composites. b Drop percentage of melting enthalpies after heated for 500 h
Conclusions
The n-OD@SiO2-PHEMA hybrid shell microcapsules are successfully prepared by interfacial hydrolysis polycondensation of alkoxy silanes and radical polymerization of HEMA, and the n-OD@SiO2-PHEMA possess the dense and smooth spherical shape. And the n-OD@SiO2-PHEMA hybrid shell microcapsules are wrapped in the network structure of PU by observing the microstructure of PU/n-OD@SiO2-PHEMA composites through SEM. The phase change enthalpies of PU/n-OD@SiO2-PHEMA composites increase with the contents of n-OD@SiO2-PHEMA. The prepared PU/n-OD@SiO2-PHEMA composites have low leakage rate and excellent mechanical properties. PU matrix and SiO2-PHEMA hybrid shell can effectively provide dual barriers to prevent the n-OD to give away even after the phase occurs at 500 h of heating environment. The tensile strength and elongation at break of the PU/n-OD@SiO2-PHEMA composites increase with the content of n-OD@SiO2-PHEMA hybrid shell microcapsules, and the tensile strength of PU/n-OD@SiO2-PHEMA can be up to 12.98 Mpa. Excellent phase change enthalpy and low hardness of the prepared form-stable PU/n-OD@SiO2-PHEMA composites indicate that they have great potential application in thermal energy storage.
References
Patil SB, Basavarajappa PS, Ganganagappa N, Jyothi MS, Raghu AV, Reddy KR (2019) Barium titanate nanostructures for photocatalytic hydrogen generation and photodegradation of chemical pollutants. Int J Hydrog Energy 44:13022–13039
Srinivas M, Reddy C, Venkata R, Kakarla RR, Shetti NP, Reddy MS, Anjanapura VR (2019) Novel co and Ni metal nanostructures as efficient photocatalysts for photodegradation of organic dyes. Mater Res Express 6:125502
Jyothi MS, Reddy KR, Naveen S, Raghu AV (2019) Nanocarbons-supported and polymers-supported titanium dioxide nanostructures as effificient photocatalysts for remediation of contaminated wastewater and hydrogen production. Nanophotocatal Environ Appl 30:83–105
Reddy KR, Jyothi MS, Raghu AV, Sadhu V, Naveen S, Aminabhavi TM (2020) Nanocarbons-supported and polymers-supported titanium dioxide nanostructures as efficient photocatalysts for remediation of contaminated wastewater and hydrogen production. Nanophotocatal Environ Appl. https://doi.org/10.1007/978-3-030-12619-3_6
Shwetharani R, Chandan HR, Sakar M, Balakrishna GR, Reddy KR, Raghu AV (2020) Recent advances in non-metals-doped TiO2 nanostructured photocatalysts for visible-light driven hydrogen production, CO2 reduction and air purification. Int J Hydrog Energy 45:18289–18308
Jinendra U, Kumar J, Nagabhushana BM, Raghu AV, Bilehal D (2019) Photocatalytic semiconductor thin films for hydrogen production and environmental applications. Green Mater 7:137–142
Karthik KV, Reddy CV, Reddy KR, Ravishankar R, Sanjeev G, Kulkarni RV (2019) Barium titanate nanostructures for photocatalytic hydrogen generation and photodegradation of chemical pollutants. J Mater Sci Mater Electron 30:20646–20653
Jinendra U, Bilehal D, Nagabhushana BM, Reddy KR, Reddy CV, Raghu AV (2019) Facile synthesis of CoFe2O4 nanoparticles and their application in removal of malachite green dye. Mater Sci Energy Technol 2:657–666
Pielichowska K, Pielichowski K (2014) Phase change materials for thermal energy storage. Prog Mater Sci 65:67–123
Zhang N, Yuan Y, Cao X, Du Y, Zhang Z, Gui Y (2018) Latent heat thermal energy storage systems with solid-liquid phase change materials: a review. Adv Eng Mater 20:1700753
Giro-Paloma J, Martínez M, Cabeza LF, Fernández AI (2016) Types, methods, techniques, and applications for microencapsulated phase change materials (MPCM): a review. Renew Sustain Energy Rev 53:1059–1075
Latibari ST, Mehrali M, Mehrali M, Afifi ABM, Mahlia TMI (2015) Akhiani AR facile synthesis and thermal performances of stearic acid/titania core/shell nanocapsules by sol–gel method. Energy 85:635–644
Geng X, Li W, Yin Q, Wang Y, Han N, Wang N (2018) Design and fabrication of reversible thermochromic microencapsulated phase change materials for thermal energy storage and its antibacterial activity. Energy 159:857–869
Geng X, Li W, Wang Y, Lu J, Wang J, Wang N (2018) Reversible thermochromic microencapsulated phase change materials for thermal energy storage application in thermal protective clothing. Appl Energy 217:281–294
Wang Y, Xia TD, Feng HX, Zhang H (2011) Stearic acid/polymethylmethacrylate composite as form-stable phase change materials for latent heat thermal energy storage. Renew Energy 36:1814–1820
Qiu X, Li W, Song G, Chu X, Tang G (2012) Microencapsulated n-octadecane with different methylmethacrylate-based copolymer shells as phase change materials for thermal energy storage. Energy 46:188–199
Alkan C, Sari A, Karaipekli A (2011) Preparation, thermal properties and thermal reliability of microencapsulated n-eicosane as novel phase change material for thermal energy storage. Energy Convers Manag 52:687–692
Yoo Y, Martinez C, Youngblood JP (2017) Synthesis and characterization of microencapsulated phase change materials with poly(urea−urethane) shells containing cellulose nanocrystals. ACS Appl Mater Interfaces 9:31763–31776
Mochane MJ, Luyt AS (2012) Preparation and properties of polystyrene encapsulated paraffin wax as possible phase change material in a polypropylene matrix. Thermochim Acta 544:63–70
Jiang Z, Yang W, He F, Xie C, Fan J, Wu J (2018) Modified phase change microcapsules with calcium carbonate and graphene oxide shells for enhanced energy storage and leakage prevention. ACS Sustain Chem Eng 6:5182–5191
Liang SE, Li QB, Zhu YL, Chen KP, Tian CR, Wang JH (2015) Nanoencapsulation of n-octadecane phase change material with silica shell through interfacial hydrolysis and polycondensation in miniemulsion. Energy 93:1684–1692
Fortuniak W, Slomkowski S, Chojnowski J, Kurjata J, Tracz A, Mizerska U (2013) Synthesis of a paraffin phase change material microencapsulated in a siloxane polymer. Colloid Polym Sci 291:725–733
Zhu Y, Qin Y, Liang S, Chen K, Tian C, Wang J (2019) Graphene/SiO2/n-octadecane nanoencapsulated phase change material with flower like morphology, high thermal conductivity, and suppressed supercooling. Appl Energy 250:98–108
Zhu Y, Qin Y, Wei C, Liang S, Luo X, Wang J (2018) Nanoencapsulated phase change materials with polymer-SiO2 hybrid shell materials: compositions, morphologies, and properties. Energy Convers Manag 164:83–92
Umair MM, Zhang Y, Iqbal K, Zhang S, Tang B (2019) Novel strategies and supporting materials applied to shape-stabilize organic phase change materials for thermal energy storage—a review. Appl Energy 235:846–873
Karaman S, Karaipekli A, Sarı A, Biçer A (2011) Polyethylene glycol (PEG)/diatomite composite as a novel form-stable phase change material for thermal energy storage. Sol Energy Mater Sol Cells 95:1647–1653
Tang B, Wang L, Xu Y, Xiu J, Zhang S (2016) Hexadecanol/phase change polyurethane composite as form-stable phase change material for thermal energy storage. Sol Energy Mater Sol Cells 144:1–6
Guo Y, Yang W, Jiang Z, He F, Zhang K, He R (2019) Silicone rubber/paraffin@silicon dioxide form-stable phase change materials with thermal energy storage and enhanced mechanical property. Sol Energy Mater Sol Cell 196:16–24
Rostami M, Ranjbar Z, Mohseni M (2010) Investigating the interfacial interaction of different aminosilane treated nano silicas with a polyurethane coating. Appl Surf Sci 257:899–904
Nam K-H, Seo K, Seo J, Khan SB, Han H (2015) Ultraviolet-curable polyurethane acrylate nanocomposite coatings based on surface-modified calcium carbonate. Prog Org Coat 85:22–30
Król P, Król B, Kozakiewicz J, Zapotoczny S, Pilch-Pitera B, Kozdra S (2015) Composites prepared from polyurethanes modified with silicone-acrylic nanopowders. Prog Org Coat 81:72–79
Wu T, Liu Y, Li N, Huang G-W, Qu C-B, Xiao H-M (2019) Cryogenic mechanical properties of epoxy resin toughened by hydroxyl-terminated polyurethane. Polym Test 74:45–56
Do T, Ko YG, Chun Y, Choi US (2015) Encapsulation of phase change material with water-absorbable shell for thermal energy storage. ACS Sustain Chem Eng 3:2874–2881
Raghu AV, Gadaginamath GS, Aminabhavi TM (2005) Synthesis and characterization of novel polyurethanes based on 1,3-bis(hydroxymethyl) benzimidazolin-2-one and 1,3-bis(hydroxymethyl) benzimidazolin-2-thione hard segments. J Appl Polym Sci 98:2236–2244
Raghu AV, Gadaginamath GS, Priya M, Seema P, Jeong HM, Aminabhavi TM (2008) Synthesis and characterization of novel polyurethanes based on N1, N4-bis[(4-hydroxyphenyl) methylene] succinohydrazide hard segment. J Appl Polym Sci 110:2315–2320
Suhas DP, Jeong HM, Aminabhavi TM, Raghu AV (2014) Preparation and characterization of novel polyurethanes containing 4,40-{oxy-1,4-diphenyl bis(nitromethylidine)}diphenol schiff base diol. Polym Eng Sci 54:24–32
Acknowledgements
The research is especially grateful for the sponsored by Support Program (Industrial) of Changzhou Science and Technology (CE20200014, CE20170029), Jiangsu Province Graduate Cultivation Innovation Project, Double ‘Ten Hundred and Thousand’ Project of Wujin District Science and Technology Bureau, the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions (TAPP), the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), Jiangsu Chenguang Paint Co., Ltd., Changzhou, Jiangsu and Shenzhen Basic Research Project (JCYJ20170818114324998).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Lin, X., Chen, Y., Jiang, J. et al. Polyurethane/n-octadecane@silicon dioxide-polyhydroxyethyl methacrylate form-stable phase change materials with enhanced mechanical properties and thermal energy storage. Polym. Bull. 79, 3867–3889 (2022). https://doi.org/10.1007/s00289-021-03677-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-021-03677-y