
Abstract
This work was focused on the synthesis of polyurethane networks (PUNs) based on polycaprolactone (PCL) as soft segment (SS) and Boltorn® aliphatic hyperbranched polyester of the second pseudo generation and 4,4′-diphenylmethane diisocyanate (MDI) as the parts of the hard segments (HS), by a two-step solution polymerization. In order to find the best ratio between HS and SS to achieve good thermal and mechanical properties, suitable hydrophobicity and morphology of the PUNs, various experimental analyses were conducted. The obtained results revealed that features of the PUNs highly depend on the PCL content. The increase in the PCL content leads to the increase in thermal stability, hydrophobicity and appearance of the microphase separation, but on the other side, it also leads to the decrease in storage modulus in the rubbery plateau, crosslinking density and glass transition temperatures of PUNs. This work shows that features of PUNs can be easily adjusted for a specific application by careful selection of the SS and HS ratio.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Segmented polyurethanes (PUs) represent a wide range of polymeric materials that have been constructed of alternating rigid urethane parts (hard segments, HS) and flexible polyol parts (soft segments, SS). By appropriate selection of reactants that includes different diisocyanates, polyols with a defined molecular weight, as well as chain extenders, it is possible to obtain PUs with different chemical and physical properties, customized for a variety of applications [1,2,3]. In order to improve their mechanical properties, thermal and chemical resistance, polyurethane networks (PUNs), can be prepared using adequate multifunctional reactants [4, 5]. PUNs are, compared to the linear PUs, polymeric materials with a three-dimensional chemically crosslinked structure which is insoluble in any solvent. The features of the PUNs depend on the degree of crosslinking, molecular weight between crosslinks (Mc), type and functionality of reactants, intermolecular forces between polymer chains, etc. [6,7,8]. Crosslinked structure of PUNs leads to the improvements in tensile strength, storage and loss modulus, as well as increase in Tg value, but on the other hand induces deterioration of their elongation at break [9].
Hyperbranched polyesters (HBPs) are multifunctional polymers (with a large number of end groups) that are often applied as crosslinking agents in the synthesis of the PUNs. HBPs have received increasing attention due to their three-dimensional and branched structure, low viscosity, good solubility and unique chemical and physical properties [10,11,12]. High functionality of the HBPs allows fast curing and formation of highly crosslinked structure of PUNs, which was applied for development of different coatings [13], adhesives [14] and elastomers [15]. For example, Manjula Dhevi et al. [16] investigated the influence of various generations (from the first to the fourth) of HBPs on the toughness of the crosslinked polyurethane/epoxy-g-interpenetrating polymer networks. Compared to the pure epoxy and linear polyurethane/epoxy samples, the crosslinked polyurethane/epoxy-g-interpenetrating networks exhibit higher impact strength, due to the existence of the two-phase morphology. Somisetti et al. [17] have used novel HBPs (degree of branching equal to 66%), obtained from renewable resource, as crosslinking agents for the preparation of poly(urethane-urea) coatings. Hydrophobicity, Tg value and crosslinking density of these PUNs increase with increasing –NCO/OH ratio. Zhou et al. [15] synthesized high-damping polyurethane elastomers based on hyperbranched polyester, which enabled increase in the free volume and degree of hydrogen bonding, decrease of microphase separation and wide effective damping temperature range.
Polycaprolactone (PCL) is synthetic aliphatic polyester which was often used as SS to prepare linear or crosslinked PUs. PCL diols, with different Mn, have low glass transition and melting temperature, good hydrophobicity and semicrystallinity, and they represent good choice to provide satisfactory thermal and surface properties when embedded in the PU chains [18]. Furthermore, PCL possess good processability, biodegradability and biocompatibility, but relatively poor mechanical and barrier properties [19]. These drawbacks can be overcome by binding PCL into crosslinked structure of the PUNs. Moreover, PCL has the ability to easily combine with other polymeric components during the preparation of different PUNs such as poly(ethylene glycol) [20], poly(propylene glycol) [21], poly(dimethylsiloxane) [22], poly(lactic acid) [23], poly(butylene succinate) [24] etc. In accordance to that Zhao et al. [22] synthesized poly(siloxane-urethane) elastomers based on PCL diol (Mn = 4000 g mol−1) as SS, in order to enhance the mechanical properties of these networks. They have shown that the obtained microphase separated morphology and crosslinked structure enabled good tensile strength, elasticity and shape memory efficiency of poly(siloxane-urethane) elastomers. Furthermore, Kupka et al. [20] used heat cured—solvent free procedure in order to obtain PUNs with HS based on hexamethylene diisocyanate and SS based on poly(ethylene glycol) (PEG) and PCL. They have shown that addition of PCL strongly improves mechanical properties of PUNs, especially when the ratio of PCL/PEG was higher than 1.8, due to the formation of crystalline PCL domains. Yuan et al. [25] prepared crosslinked PUN binders based on hydroxy-terminated polyether (HTPE) (Mn = 3190 g mol−1) and PCL (Mn = 2000 g mol−1), by changing the relative mass content of PCL to HTPE. They have found that with increasing PCL content the degree of microphase separation decreases, leading to the improvement of mechanical properties (PUN with 40 wt% of PCL), especially at low temperatures. Besides, Unal et al. [26] have shown that it is possible to use trifunctional PCL as the SS for the synthesis of the PUNs. They obtained highly branched networks with slightly better mechanical properties compared to the linear analogue. Furthermore, they have demonstrated that PCL segment was amorphous in highly branched PUNs, while it showed a certain degree of crystallinity in the linear analogue PCL with equivalent molecular weight. In our previous work, we prepared a series of PUNs based on PCL, Boltorn® hyperbranched polyester and isophorone diisocyanate in order to investigate the influence of the PCL content on different properties of these PUNs [27]. The increase in the PCL content caused appearance of the microphase separated morphology, and led to the increase in thermal stability, elongation at break, hydrophobicity and swelling ability in tetrahydrofuran and toluene. On the other hand, PUNs with higher PCL content showed lower crosslinking density, glass transition temperatures, tensile strength and Young’s modulus [27].
In this work, we have extend our investigation of PUNs and prepared a new series of PU networks based on PCL diol (as SS) and Boltorn® hyperbranched polyester of the second pseudo generation (BH-20) and MDI (as parts of HS). The aim was to check the properties and the quality of the prepared PUNs when 4,4′-diphenylmethane diisocyanate is applied instead of isophorone diisocyanate. A catalyzed two-step polymerization technique in a solvent mixture of DMAc/THF was used. Six PUN films with a content of soft PCL segment between 10 and 60 wt% were synthesized. The influence of different composition (HS/SS ratio) on the structure, thermal and mechanical properties, as well as on the morphology and swelling behavior of prepared PUNs was tested by different experimental techniques. The novelty of this work is reflected in several terms. First, to the best of our knowledge, there are only a few researches dealing with the PUNs based on some of these reactants. There are no reports on this exact combination of reactants (BH-20, PCL diol with Mn ≈ 2000 g mol−1, MDI) which assemble their properties, particularly those important for application in coatings. Second, the comparison of performances of PUNs prepared with isophorone diisocyanate in our previous work [27] with PUNs prepared with MDI in the present work gave valuable information, especially those considering easiness of tuning thermal (thermal stability, glass transition temperature) and mechanical properties (tensile strength, Young’s modulus) of PUNs by changing applied diisocyanate. And third, in the present work, we have extended our investigation of swelling properties of PUNs, by a detailed examination of swelling kinetics and diffusion mechanism of solvents and calculation of diffusion coefficients for PUNs.
Experimental section
Materials
Polycaprolactone diol (PCL, Mn ≈ 2000 g mol−1) was supplied from Sigma-Aldrich (purity ≤ 100%) and dried at 80 °C for 12 h prior to use. Boltorn® hydroxy-functional aliphatic hyperbranched polyesters of the second pseudo generation (BH-20) was supplied from Perstorp Specialty Chemicals AB and dried under vacuum for 48 h at 50 °C before application. Functionality of BH-20 was calculated (f = 12) from the Mn determined by vapor pressure osmometry (Mn = 1340 g mol−1), and hydroxyl number determined by titration method was 501.1 mg KOH g−1 [28]. 4,4′-Diphenylmethane diisocyanate (MDI) and toluene was supplied from Sigma-Aldrich (purity 98% and 99.9%, respectively) and used as received. The catalyst solution stannous octoate (Sn(Oct)2) (supplied from Sigma-Aldrich, purity 95%) was prepared by dissolving an appropriate amount of Sn(Oct)2 in an anhydrous N,N-dimethylacetamide (DMAc). DMAc, purchased from Sigma-Aldrich (purity 99%), was distilled over CaH2 under reduced pressure, while tetrahydrofuran (THF), obtained from J. T. Baker (purity 99.99%), was distilled over LiAlH4. Both solvents were kept over molecular sieves (0.4 nm) before the synthesis.
Synthesis
The series of PUN samples based on PCL, MDI and BH-20 were prepared by a two-step polymerization in solution, using a mixture of DMAc/THF as solvent and Sn(Oct)2 as a catalyst. The total molar ratio of –NCO/OH groups was kept constant at 1.05 for all prepared PUNs.
The detailed reaction procedure for the synthesis of PUNMDI-40 sample was as follows: the desired amounts of PCL prepolymer (3.00 g, 1.50 mmol) and MDI (0.79 g, 3.15 mmol) were dissolved in THF (24.10 ml) in a four-necked round-bottom flask, equipped with mechanical stirrer, dry argon inlet, reflux condenser and a dropping funnel and placed in an oil bath heated up to 60 °C. After addition of catalyst solution (0.15 mL, 0.15 mol% of Sn(Oct)2 with regard to the PCL prepolymer) into the reaction mixture, the polymerization reaction started. The reaction mixture was continuously stirred at 60 °C for 2 h in order to obtain NCO-terminated prepolymer, which was monitored using the standard dibutylamine back-titration method [29]. In the second step of the polymerization reaction, a dilute solution of BH-20 (1.89 g, 1.41 mmol) in DMAc (6.70 ml) and excess of MDI (1.83 g, 7.30 mmol) in DMAc/THF (4.00/5.00 ml) were dropwise added to the flask and the reaction mixture was additionally stirred for 15 min at 60 °C. The PUN films were obtained after pouring the content of the reaction mixture into the Teflon dishes and curing at 40 °C in a force-draft oven for 26 h. After that PUN films were dried under vacuum at 50 °C for 24 h and then kept in desiccators for 14 days at room temperature before characterization. Other samples were prepared in the same manner, taking into account the desired SS content in the final polymer. Six samples of the synthesized PUNs were labeled as PUNMDI-10, PUNMDI-20, PUNMDI-30, PUNMDI-40, PUNMDI-50 and PUNMDI-60, where the numbers represent the weight percent of the SS.
Characterization
FTIR spectroscopy
Infrared spectra were taken in ATR mode using a Nicolet 6700 FTIR spectrometer. During recording, the conditions for each PUNs were as follows: resolution of 2 cm–1, number of scans 64 and interval 400–4000 cm–1.
X-ray diffraction technique
X-ray diffraction (XRD) measurements were conducted on an Ital Structure APD 2000 X-ray powder diffractometer with CuKα1 radiation (λ = 0.15418 nm), where step-width and step-time for all PUNs were 0.02° and 1 s, respectively. The data were collected in the 2θ range from 15° to 35° on the PUNs, which dimensions were 25 mm × 25 mm × 1 mm.
Swelling measurements
Swelling of PUNs was investigated in THF and toluene at room temperature. Square test samples (10.0 mm × 10.0 mm × 1.0 mm ± 0.2 mm) were immersed in selected medium, and their weight, w, was periodically measured until constant value, i.e., equilibrium swelling. The excess of medium was before each weight measurement carefully removed with filter paper. The swelling degree, q, was estimated as follows:
where w0 is the weight of PUN before immersion in THF or toluene. Results obtained from three separate measurements were averaged.
The crosslinking density of the PUNs, i.e., molar number of elastically effective network chains per cm3, ν, was calculated from the results obtained by separate swelling behavior investigation of PUNs in THF and Flory-Rehner equation for equilibrium swollen network [30]:
where VPUN represents volume fraction of the crosslinked polymer in the swollen specimen, χ is the polymer–solvent interaction parameter, and Vs is the molar volume of the THF (81.7 cm3 mol−1) [31]. VPUN can be calculated using the following equation:
where ρPUN represents the density of PUNs (Table 1), ρs is the density of THF (ρs = 0.889 g cm−3) [31], w is the weight of the swollen PUN measured after being soaked in THF for 48 h, while wg is the weight of the deswollen sample measured after drying swollen sample in vacuum oven at 50 °C until the weight reached constant value. Polymer–solvent interaction parameter was calculated using values for the solubility parameters of PUNs, δPUN, determined from the Hoy molar attraction constants, and solubility parameters of THF (δTHF = 18.6 J1/2 cm−3/2) [31]:
where R is the gas constant and T is the temperature. Finally, the number average molecular weight of polymer chain between crosslinks, Mc, can be calculated as follows:
Values of the Mc calculated in this manner are number averaged and not dependent on the crosslinks distribution. Finally, the gel content was calculated from the values of w0 and wg in the following manner:
Each value (VPUN, ν, Mc and gel %) represents the average of three repeated experiments.
Density measurements
Density of PUNs was determined using pycnometer and distilled water as medium at room temperature. Results obtained from three separate measurements were averaged.
Water absorption measurements
The weight percent of the water absorption, WA, of PUNs was determined by the immersion of square test samples (10.0 mm × 10.0 mm × 1.0 mm ± 0.2 mm) in distilled water at room temperature for 48 h. Values of WA were calculated using the following equation:
where wW0 is the weight of dry sample, and wW is the weight of PUN after immersion in distilled water for 48 h. The average of three measurements was used.
Thermogravimetric analysis
Thermogravimetric analysis (TG) was performed using an SDT Q600 (TA Instruments) thermogravimetric analyzer. The samples were heated from 20 to 700 °C at a heating rate of 10 °C/min and in nitrogen flow (0.1 dm3 min−1).
Differential scanning calorimetry
Differential scanning calorimetry (DSC) analysis was performed on a Netzsch DSC 204 F1 Phoenix® instrument. The samples were analyzed in the temperature range from − 80 to 180 °C, at a heating rate of 10 °C min−1 and in nitrogen flow of 20 mL min−1. Values of glass transition temperature of the soft and hard segments of the prepared PUNs were determined as the midpoint of the glass transition event.
Dynamic mechanical analysis
Dynamic mechanical analysis (DMA) of PUNs was done on Perkin Elmer Pyris Diamond DMA analyzer using the film tensile configuration. Measurements were performed on rectangular PUN samples (dimensions 20 mm × 5 mm × 0.5 mm ± 0.1 mm), at a frequency of 1 Hz, strain amplitude of 5 μm, in the temperature range from − 100 to 175 °C and with a heating rate of 3 °C min−1. The crosslinking density of the PUNs was calculated using the DMA results and the following equation based on the rubber-like elasticity [32, 33]:
where E′ represents storage modulus, R is the universal gas constant, and T is the absolute temperature. Values of the Mc of the investigated PUNs were calculated using the Eq. 5.
Tensile tests
Mechanical properties of dumbbell-shaped PUN samples (dimensions 60 mm × 10 mm × 0.5 mm) were examined using an AG–X plus Universal Testing Machine, Shimadzu. Mechanical properties were tested at room temperature and with crosshead speed of 5 mm min−1. The obtained results are the mean values of three repetitions for each sample.
SEM analysis
SEM microphotographs of PUN sample surfaces and cross-sections were obtained on a JEOL JSM-6460LV scanning electron microscope. Measurements were carried out at a working distance of ca. 14 mm and with the accelerating voltage of 20 kV. Before the SEM recording, PUN samples were cryo-fractured in liquid N2 and coated with a thin Au layer.
Results and discussion
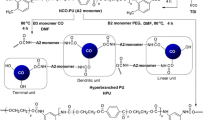
A series of six PUNs with various SS content, ranging from 10 to 60 wt%, was successfully prepared in order to examine the influence of SS content on the morphology, surface, thermal and mechanical properties of these networks. The presence of hyperbranched polyester (BH-20) as crosslinking agent with its 12 end hydroxyl groups enabled fast curing of the polyurethane films. Figure 1 shows the chemical structures of reactants, schematic representation of the two-step polymerization reaction and photographs of the prepared PUN films. According to the presented photographs (Fig. 1c), the transparency of the obtained PUNs slightly decreases with increasing PCL content, probably as a consequence of the crystallization of PCL in the PUNMDI-50 and PUNMDI-60 samples.

a Chemical structure of the reactants, b schematic representation of the two-step polymerization reaction and c photographs of the synthesized PUN films
Results of FTIR analysis
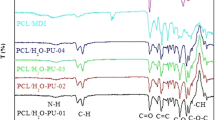
FTIR analysis was performed to check the chemical structure of the synthesized PUNs. Characteristic bands of different groups present in crosslinking agent (BH-20), PCL prepolymer and selected PUN samples are visible in the FTIR spectra (Fig. 2).

FTIR spectra of BH-20 crosslinking agent, PCL prepolymer and selected PUN samples
FTIR spectrum of BH-20 shows broad stretching vibrations of –OH groups (3300 cm−1), stretching vibrations of –CH2 and –CH3 groups (2880–2940 cm−1), stretching vibrations of –C = O groups (1723 cm−1) and stretching vibrations of C–O bonds of ester groups (1210 cm−1) and ether groups (1010–1115 cm−1). In the FTIR spectrum of PCL prepolymer, stretching vibrations of –OH groups (around 3500 cm−1), stretching vibrations of symmetric and asymmetric–CH2 groups (2868 and 2950 cm−1, respectively), stretching vibrations of –C = O groups (1726 cm−1), stretching vibrations of asymmetric and symmetric C–O–C (1242 and 1186 cm−1, respectively), stretching vibrations of C–O and C–C groups in the crystalline and amorphous phase (1295 and 1167 cm−1, respectively) and deformation vibrations of –CH2 groups (1370 cm−1) can be observed.
FTIR spectrum of synthesized PUNs shows distinct bands related to stretching vibrations of C–O ether and C–O ester groups (1063 and 1166 cm−1, respectively), stretching vibrations of aromatic C = C groups (1624 and 1415 cm−1), stretching vibrations of amide II and III groups (1540 and 1232 cm−1, respectively), stretching vibrations of –C = O groups (1732 cm−1), stretching vibrations of asymmetric and symmetric –CH2 and –CH3 groups (2945, 2865 cm−1, respectively), stretching vibrations of N–H bonds (3320 cm−1), as well as stretching vibrations of free –OH groups (3740 cm−1). The absence of –NCO band at 2260 cm−1 and the presence of carbonyl and N–H bands indicate complete conversion of the –NCO groups into urethane bonds. From Fig. 2 one can see that characteristic bands of N–H, aromatic, urethane carbonyl, amide II and III, and C–O ether groups are quite pronounced and more intense in the FTIR spectrum of samples with higher HS content. Higher intensity and existence of wider bands characteristic for N–H and carbonyl urethane groups may indicate higher level of hydrogen bonding between these groups in PUN samples with higher HS content. According to the literature, a higher level of hydrogen bonding usually leads to the PUNs with good mechanical properties [34, 35]. The obtained FTIR spectra confirm successful preparation of PUN samples with different compositions i.e. HS/SS ratio.
Results of XRD analysis
XRD analysis was used to analyze the crystal structure of the PCL prepolymer and synthesized PUNs. Figure 3 shows the XRD patterns of PCL prepolymer and PUNs.

XRD patterns of PCL prepolymer and PUN samples
In the XRD pattern of neat PCL prepolymer, there are three intense reflections of the (110), (111) and (200) planes at 2θ = 21.5°, 22.1° and 23.9°, respectively. These reflections indicate the existence of the orthorhombic crystal lattice of semicrystalline PCL prepolymer [36, 37]. On the contrary, a broad and weak reflection at 2θ ~ 20.2° are the consequence of amorphous contribution [37]. Three reflections, typical for the neat PCL, are also visible in the XRD pattern of PUNMDI-60, but less intense. However, in all diffractograms, except for PUNMDI-10, also weaker reflections at 2θ = 21.5° and 23.9°, as well as broad reflection at 2θ ~ 20.2°, are vaguely visible. The reflection at 2θ = 22.1° is not expected to be seen in PUNs, since it is the weakest one in PCL. The existence of less intense and wider reflections in the XRD pattern of PUNs with lower PCL content and total absence of reflections in PUNMDI-10 sample indicates that PCL chains were not able to crystallize due to the presence of polymer network [27]. As a consequence of that these PUNs are amorphous.
Results of swelling and water absorption measurements
The swelling behavior of PUNs was investigated by determination of their equilibrium swelling degree, qe, in toluene and THF (Fig. 4a). Swelling ability of these PUNs increases with increasing SS content in both solvents and it is higher in THF than in toluene, since THF solvates prepared networks better than toluene. Because of that the extent of chemical crosslinking of prepared PUNs was estimated from the results obtained by swelling of samples in THF for 48 h. Using the Eqs. 2–6, values of the volume fraction of crosslinked polymer in the swollen specimen, VPUN, crosslinking density, ν, the number average molecular weight of polymer chain between crosslinks, Mc and the gel content of prepared PUNs was calculated and summarized in Table 1. The obtained results show that with decreasing SS content values of VPUN, ν and gel content increase, while Mc decreases due to the presence of higher amount of crosslinking agent BH-20 during the synthesis of PUNs. In addition, values of the crosslinking density obtained for these networks are of the same order of magnitude as those obtained for other PUNs based on the same crosslinking agent [4, 38, 39].

a The dependence of the equilibrium swelling degree, qe, determined in THF and toluene and b percent of the water absorption of PUNs on the SS content
The percent of the water absorbed by PUNs after their immersion in distilled water for 48 h at room temperature are given in Fig. 4b. The hydrophobicity of these PUNs increases with increasing SS content, probably due to the hydrophobic character of PCL [40,41,42].
In order to study swelling kinetics of the prepared PUNs, the pseudo-first-order and pseudo-second-order kinetic model were applied to analyze experimental data [43, 44]. The pseudo-first-order model can be expressed with the following equation:
while the pseudo-second-order model can be formulated in the following manner:
where qt = wt/w0 represents swelling degree at time t, wt (g) is the amount of solvent (toluene, THF or water) penetrated into PUN at time t and w0 is the weight of PUN before swelling experiment. Furthermore, qe1 and qe2 are theoretical equilibrium swelling degrees calculated from curve-fitting using pseudo-first-order and pseudo-second-order model, respectively, and k1 (min−1) and k2 (g g−1 min−1) represent rate constants of the pseudo-first-order and pseudo-second-order model, respectively. The criteria usually applied to choose appropriate model for description of the swelling kinetics of polymers is based on the value of correlation coefficient (R2). Figure 5 shows curve-fitting results obtained using pseudo-second-order model for PUNs in toluene and THF, while selected values of rate constants, theoretical equilibrium swelling degrees and correlation coefficients of two swelling kinetic models of PUNs in different solvents are listed in Table 2.

Pseudo-second-order swelling kinetic model for PUNs in a toluene and in b THF
Swelling process of PUNs in toluene and THF fits more closely with the pseudo-second-order kinetic model (Fig. 5), since correlation coefficients of the pseudo-second-order model are for PUNs in these two solvents closer to 1.0 than R2 of the pseudo-first-order model. Also, values of qe2 are in much better agreement with the experimental qe than qe1. Therefore, the swelling process of PUNs in toluene and THF can be well described with pseudo-second-order kinetic model. For sample PUNMDI-10 in toluene the fitting curve has bigger slope than curves for other PUNs, because qe2 is for this network one order of magnitude lower than qe2 of other PUNs swollen in toluene (Table 2). The pseudo-first-order model is more suitable than pseudo-second-order kinetic model for describing swelling process of PUNs in water, since values of qe1 agree well with experimental qe and R2 of the pseudo-first-order model is higher than 0.99. The increase of k2, and consequently the increase of swelling rate, with increasing SS content up to the 50 wt% in toluene and up to the 40 wt% in THF is probably caused by the decrease in the crosslinking density of PUNs. The decrease in swelling rate with further SS content increase is caused by the microphase separation in PUNs with higher SS content. Namely, depending on their size, shape and distribution, hard segments can alter the path of the solvents molecules and consequently slow down the transport of solvent through the network [45]. Furthermore, results given in Table 2 show that swelling rate of PUNs in toluene is higher than in THF, which is consistent with the experimentally obtained results. During swelling measurements, equilibrium swelling degrees of PUNs were reached faster in toluene (after 30 h) than in THF (after 48 h). Different values of k1 indicate different water absorption rates of PUNs, but generally it can be concluded that water absorption rate is higher for PUNs with lower crosslinking density.
The common manner to investigate the diffusion mechanism of solvents into the polymer is based on simple power-law expression [43, 46]:
where we (g) is the amount of solvent penetrated into PUN when equilibrium swelling was reached, k is the power-law constant, and n is the diffusion exponent which describes the type of the diffusion mechanism and can be determined as slope of the dependence log (wt/we) vs. log t. Given equations are valid only for the wt/we ≤ 0.6, i.e. below 60% of the total equilibrium weight uptake [46]. Since for PUNs investigated in this work wt/we ≤ 0.6 was obtained only for three samples swollen in toluene (PUNMDI-10, PUNMDI-20 and PUNMDI-30) and all samples swollen in water, Fig. 6 shows the dependence of the diffusion exponent n on the SS content only for these PUNs.

The dependence of the diffusion exponent n on the SS content for PUNs in toluene and water
It is well known that the transport of solvent through a polymer happens when solvent molecules pass through the gaps between polymer segments. Due to that the diffusion rate depends on the size of solvent molecules and size of gaps between polymer segments, while the size of gaps depends on physical properties of polymer. Taking that into account, Alfrey et al. proposed three cases which can be used to describe transport phenomena of solvent through the polymer, based on the solvent diffusion rate (Rdiff) and the polymer relaxation rate (Rrelax) [47]. Case I represents Fickian diffusion, characterized by Rdiff < < Rrelax. This case is common for polymer networks at temperatures well above glass transition temperature of the polymer, where the mobility of polymer chains is higher, which facilitates solvent penetration into the polymer. For case I type of solvent transport through the thin polymer films, the diffusion exponent (eqs. (11) and (12) is n = 0.5. Non-Fickian diffusion (case II) is characterized by Rdiff > > Rrelax, and it is mostly observed in polymer networks at temperatures below glass transition temperature, where the mobility of polymer chains is low and not sufficient to allow fast solvent penetration. For case II type of transport and thin polymer films n is equal to 1.0. The case III also represents non-Fickian diffusion, called anomalous diffusion, which is characterized by Rdiff ≈ Rrelax, when both polymer relaxation and solvent diffusion control the rate of solvent penetration. For this case, the diffusion exponent is 0.5 < n < 1.0. There are also 2 other diffusion mechanisms classified depending on the value of n: pseudo-Fickian diffusion (n < 0.5) and super case II diffusion (n > 1.0) [43]. According to the results given in Fig. 6, diffusion exponent is only for PUNMDI-10 in the range 0.5 < n < 1.0, indicating that both polymer relaxation and solvent diffusion control swelling rate of this network in toluene and water. For all other PUNs given in Fig. 6, the toluene and water transport mechanism is pseudo-Fickian (n < 0.5).
The diffusion coefficients (D) for the prepared PUNs were calculated from the swelling data. For diffusion process which occurs at short times (wt/we ≤ 0.5), i.e., for all samples in water and PUNMDI-10, PUNMDI-20 and PUNMDI-30 in toluene, the following equation was applied [48]:
where L is the thickness of sample (1.0 mm ± 0.2 mm). From the slope of the linear part of dependence wt/we vs. t1/2/L, value of D was calculated and listed in Table 3. For long time diffusion and wt/we ≥ 0.5 (all samples in THF and PUNMDI-40, PUNMDI-50 and PUNMDI-60 in toluene) it is more accurate to use Eq. (14) for the calculation of D [48, 49]:
In this case, D was calculated from the slope of dependence ln (1− wt/we) vs. t and obtained values are summarized also in Table 3. Diffusion coefficient for prepared PUNs increases with decreasing crosslinking density, i.e., with increasing SS content up to the 40 wt% in toluene and THF and 50 wt% in water, due to the increase in the solvent ability to move along the polymer segments. In PUNs with higher SS content the diffusion of solvents is somewhat slower, probably due to the existence of microphase separation in PUNs with higher SS content.
Results of TG analysis
Thermal stability of the PUN samples was investigated using TG analysis under N2 atmosphere. TG and DTG curves of PUNs are presented in Fig. 7, while characteristic temperatures of the thermal degradation (T5, T10, T50 and T90 at 5, 10, 50 and 90% weight loss, respectively) are listed in Table 4. According to the results presented in Fig. 7a and Table 4, the beginning of thermal degradation (T5% value) of sample PUNMDI-10 was above 140 °C. On the other hand, the initial decomposition temperature of PUNMDI-60 was 264 °C, which is much higher than T5% of PUNMDI-10. This indicates that thermal stability of the prepared PUNs can be significantly improved by increasing SS content. Namely, networks with higher PCL content have simultaneously lower amount of thermally unstable urethane bonds, which is the reason for the improvement of thermal stability of these PUNs. The same was observed for PUNs based on PCL, BH-20 and isophorone diisocyanate [27]. Besides, PUN samples with higher SS content have usually better microphase separation, which is also one of the prerequisites for the thermal stability enhancement [50]. Furthermore, the obtained results also revealed that thermal stability of networks based on PCL and BH-20 could be easily tuned by choosing adequate diisocyanate. Namely, thermal stability of these PUNs is better if isophorone diisocyanate is used during the synthesis [27].

a TG and b DTG curves of the PUN samples, determined in nitrogen atmosphere, at a heating rate of 10 °C min−1
Taking into account presented DTG curves (Fig. 7b) and temperatures of DTG peaks summarized in the Table 4, it can be concluded that PUN samples have several thermal degradation steps. The first, very small peak, which can be observed in the range from 160 to 170 °C, is associated with the loss of volatile compounds, such as residual solvent. Networks with higher HS content have also higher crosslinking density and consequently higher possibility to trap larger amount of solvent than samples with higher SS content. Therefore, with increasing SS content this DTG peak decreases, and for the sample PUNMDI-60 it is not visible at all. The DTG peak which appears between 295 and 304 °C is connected to the decomposition of the urethane bonds in the HS and its intensity strongly decreases with increasing SS content [51]. This degradation step is visible only as a shoulder in DTG curves of PUNs with SS content higher than 20 wt%. Same peak has also one shoulder on its left side (around 253 °C), which is assigned to the beginning of the thermal decomposition of the HS and it has the same trend with increasing SS content as the main peak. The next decomposition step, for which DTG peak appears between 328 and 368 °C, is associated to the thermal degradation of PCL [52]. The maximum thermal degradation rate, connected to this decomposition step, considerably increases with increasing SS content and for sample PUNMDI-60 the corresponding DTG peak is therefore very intense and sharp (Fig. 7b). On the other hand, for the sample PUNMDI-10 this decomposition step had the lowest maximum thermal degradation rate due to the smallest PCL content. Besides, DTG peak of PUNMDI-10 overlaps with the next one and is shifted to the higher temperature. DTG peak associated to the thermal degradation of PCL has also one shoulder on its right side (between 400 and 424 °C), originating from the thermal degradation of the SS in the PUN samples [20, 52, 53]. According to the literature, decomposition of the urethane bonds produces isocyanates and alcohols, while the decomposition of ester and ether bonds from PCL and hyperbaranched polyester occurs through chain scission [20, 54].
Results of DSC analysis
Thermal properties of the synthesized PUNs were also investigated by DSC analysis. The obtained DSC results gathered from the second heating run are presented in the Fig. 8 and Table 5. All PUN samples have glass transition temperature of HS (TgH), while only samples with the highest SS content (PUNMDI-50 and PUNMDI-60) have also glass transition temperature of the SS (TgS). Therefore, according to the DSC results, only networks PUNMDI-50 and PUNMDI-60 have microphase separated morphology. Value of TgH of the prepared PUNs is in the range from 11 to 16 °C, and it increases with decreasing SS content, due to the increase of crosslinking density and consequent restriction of polymer chains movements. Moreover, from Fig. 8 it can also be observed that this transition is very wide, which is probably a consequence of the diverse network structure, i.e., existence of different HS lengths. Sample PUNMDI-60 has slightly lower TgS than PUNMDI-50, which indicates that TgS decreases with increasing SS content.

DSC thermograms of the PUNs
In the DSC, thermogram of PUNMDI-60 (sample with the highest PCL content), one endothermic peak at 41 °C was also observed. This endothermic peak is attributed to the melting of the SS (TmS). According to the literature, the melting temperature of the neat PCL is around 60 °C [55], while TmS value obtained in this case is lower, as a consequence of lack of organization of SS, i.e., due to the presence of crosslinked structure [27]. The presented DSC results of investigated PUNs are in accordance with the obtained XRD results.
DMA results
The influence of the SS content on the viscoelastic properties of prepared PUNs was investigated by DMA and obtained results are presented in Fig. 9 and Table 6. The DMA results of PUN sample with the highest SS content (PUNMDI-60) are not presented, since this sample broke down, i.e., it melted down, before finishing the experiment. According to the temperature dependences of the storage modulus, E′, and tan δ of other prepared PUNs it can be concluded that only samples with higher SS content have microphase separated morphology (Fig. 9). Namely, PUNs with SS content of 10 and 20 wt% (PUNMDI-10 and PUNMDI-20) show only one temperature transition corresponding to the glass transition temperature of the HS. This indicates that in PUNMDI-10 and PUNMDI-20, SS domains are dispersed and mixed in the HS, leading to the phase mixing. On the other hand, samples with higher SS content exhibit also TgS, corresponding to the transition which appears at lower temperature. Since DSC analysis enabled determination of TgS only for samples PUNMDI-50 and PUNMDI-60, it was again confirmed that DMA is the best method for determination of Tg of PUNs based on hyperbranched polyesters [27, 39]. Values of the TgS and TgH, determined from tan δ peak position, are listed in Table 6. Glass transition temperature of HS increases with SS content decrease as a consequence of the increase in the crosslinking density and rigidity of the network, while there is no specific trend for the TgS. Networks PUNMDI-10 and PUNMDI-20 have the same value of TgH, which is probably caused by the increase of the SS and HS miscibility. Furthermore, the height of tan δ maximum, corresponding to the TgS, decreases with SS content decrease, because of lower PCL content and restriction of the polymer chains mobility.

Temperature dependences of a storage modulus, E′, and b tan δ of the prepared PUNs
In the HS glass transition region (Fig. 9a), the mobility of polymer chains significantly increases compared to the ones at lower temperatures, which is the reason for the prompt E′ decrease for all investigated PUNs. Furthermore, at T > 120 °C (rubbery plateau) the existence of chemical crosslinks restricts the flow of the polymer. From the results presented in Fig. 9a, it can also be observed that network PUNMDI-10, which has the lowest SS content, has the smallest difference in E′ values below and above glass transition of HS. Also, PUNMDI-10 has the highest E′ value in the rubbery plateau, indicating that this sample has the highest crosslinking density. The effect of network composition differences on the crosslinking density can be clearly seen from the calculated values of ν and Mc of PUNs listed in Table 6. As expected, the crosslinking density of prepared PUNs decreases, while Mc increases with increasing SS content. If these results are compared with results summarized in Table 1, it can be seen that crosslinking densities of PUNs determined using DMA results are higher than crosslinking densities calculated from the swelling measurements. The opposite can be observed for the Mc values. The reason for this lies in the fact that crosslinking density evaluated from the swelling measurements considers the presence only of the chemical crosslinks, while in the case of DMA measurements the presence of both chemical and physical crosslinks are took into account. It can also be observed that values of ν have the same trend as values of the gel content determined by swelling measurements (Table 1).
Results of tensile tests
Figure 10 shows the stress–strain curves of PUNs with different SS content. Determined values of tensile strength, elongation at break and Young’s modulus, calculated from the initial linear part of the stress–strain curve, are listed in Table 7. These results show that tensile properties of PUNs are very sensitive to the SS content. The increase in SS content induced increase in the tensile strength and Young’s modulus up to the sample PUNMDI-30, indicating the simultaneous increase in the stiffness and rigidity. The reason for this can be the absence of the microphase separation in samples with lower SS content. However, further increase in the SS content reduced tensile strength and Young’s modulus of PUNs, which is probably caused by the increase in Mc and decrease in chain rigidity [4, 27, 56, 57]. Furthermore, the obtained results also show that network with the highest SS content (PUNMDI-60) has noticeable different mechanical properties than other samples, probably due to considerably lower strength of this network when subjected to the tensile test compared to other prepared PUNs. Tensile properties of PUNs synthesized in this work are quite different than results obtained for PUNs based on BH-20, PCL and isophorone diisocyanate [27]. Beside different trend of the dependence of tensile properties on SS content, PUNs prepared with isophorone diisocyanate had also higher tensile strength and considerably higher Young’s modulus [27]. On the other hand, PUNs prepared in this work showed higher values of the elongation at break than values obtained for PUNs based on isophorone diisocyanate [27]. According to these results, it can be concluded that mechanical properties of PUNs can be easily tuned not only by the SS content change, but also by adjusting the type of diisocyanate used during the synthesis of PUNs.

Stress–strain curves of PUNs
Results of SEM analysis
Surface and cross-sectional characterization of the PUN films were done by SEM analysis. Figure 11 shows SEM microphotographs of the surface and cross section of PUNs with SS content of 10, 30 and 50 wt%. According to the obtained SEM micrographs, PUNs with lower SS content have relatively smooth, uniform and homogeneous surface morphology, with little brighter circles that originate from the soft PCL segments. PUN with SS content of 50 wt% has rougher and heterogeneous surface morphology, indicating the presence of microphase separation in samples with higher SS content [58]. Cryo-fracture surfaces of PUNs presented in SEM microphotographs (Fig. 11) have irregular and flake-like structure, indicating brittle type of fracture [59, 60]. Similar morphology was reported for the polyurethane networks based on isophorone diisocyanate [27].

SEM micrographs of the surface of a PUNMDI-10, b PUNMDI-30 and c PUNMDI-50 and cross section of d PUNMDI-10, e PUNMDI-30 and f PUNMDI-50
Conclusions
In this work, a series of PUN samples with different HS/SS ratio were prepared by a two-step polymerization in solution, based on PCL, Boltron® hyperbranched polyester and MDI. FTIR analysis confirmed the chemical structure of PUN samples. Water absorption measurements showed that PUNs with higher SS content have hydrophobic nature that originates from hydrophobic PCL prepolymer. Furthermore, it has been determined that swelling ability of PUNs in toluene and THF increases with SS content increasing. The swelling process of PUNs in toluene and THF can be well described with pseudo-second-order kinetic model, while the pseudo-first-order model is more suitable for describing swelling process of PUNs in water. Diffusion coefficients for PUNs increase with decreasing crosslinking density, i.e., with increasing SS content up to the 40 wt% in toluene and THF and 50 wt% in water, due to the increase in the solvent ability to move along the polymer segments. On the other hand, the diffusion of solvents is somewhat slower in PUNs with higher SS content, due to the microphase separation in PUNs with higher SS content. XRD and DSC analyses showed that all PUNs are amorphous, except sample with the highest SS content. TG analysis revealed that incorporation of the PCL chains inside the polymer network structure increases thermal stability of the prepared PUN samples. Results obtained by swelling measurements and DMA both confirmed that the crosslinking density of prepared PUNs decreases, while number average molecular weight of polymer chain between crosslinks increases with SS content increasing. Observations gathered from DSC analysis and DMA pointed out that only samples with higher SS content have microphase separated morphology. Also, these characterization methods revealed that the glass transition temperature of HS increases with crosslinking density increase. Tensile properties of the synthesized PUNs significantly depend on the SS content and the type of diisocyanate applied during the preparation of samples. Finally, SEM analysis revealed that PUNs with higher SS content have rougher and heterogeneous surface morphology, indicating the presence of microphase separation.
The results obtained in this work show that thermal, mechanical and surface properties of the PUNs based on PCL and hyperbranched polyester can be easily adjusted for various applications by changing HS/SS ratio.
References
Fenfen W, Shengli C, Qiang W, Rongchun Z, Pingchuan S (2019) Strain-induced structural and dynamic changes in segmented polyurethane elastomers. Polymer 163:154–161
Špírková M, Poręba R, Pavličević J, Kobera L, Baldrian J, Pekárek M (2012) Aliphatic polycarbonate-based polyurethane elastomers and nanocomposites. I. The influence of hard-segment content and macrodiol-constitution on bottom-up self-assembly. J Appl Polym Sci 126:1016–1030
Yildirim E, Yurtsever M, Wilkes GL, Yilgör I (2016) Effect of intersegmental interactions on the morphology of segmented polyurethanes with mixed soft segments: a coarse-grained simulation study. Polymer 90:204–214
Maji PK, Bhowmick AK (2009) Influence of number of functional groups of hyperbranched polyol on cure kinetics and physical properties of polyurethanes. J Polym Sci Part A Polym Chem 47:731–745
Cao Q, Liu P (2006) Hyperbranched polyurethane as novel solid–solid phase change material for thermal energy storage. Eur Polym J 42:2931–2939
Okrasa L, Czech P, Boiteux G, Méchin F, Ulanski J (2007) Studies of molecular dynamics in polyurethane networks with hyperbranched polyester Boltorn®H30 as a crosslinker. J Non-Cryst Solids 353:4293–4297
Zajac M, Kahl H, Schade B, Rödel T, Dionisio M, Beiner M (2017) Relaxation behavior of polyurethane networks with different composition and crosslinking density. Polymer 111:83–90
Barszczewska-Rybarek I (2017) The role of molecular structure on impact resistance and bending strength of photocured urethanedimethacrylate polymer networks. Polym Bull 74:4023–4040
Pusztai E, Cs K, Nagy J, Wagner Ö (2013) The effect of some disiloxane chain extenders on the thermal and mechanical properties of cross-linked poly(siloxane-urethane)s. Express Polym Lett 7:456–470
Irfan M, Seiler M (2010) Encapsulation using hyperbranched polymers: From research and technologies to emerging applications. Ind Eng Chem Res 49:1169–1196
Fréchet JMJ, Hawker CJ (1995) Hyperbranched polyphenylene and hyperbranched polyesters: new soluble, three-dimensional, reactive polymers. React Func Polym 26:127–136
Bhat SI, Ahmadi Y, Ahmad S (2018) Recent advances in structural modifications of hyperbranched polymers and their applications. Ind Eng Chem Res 57:10754–10785
Kumari S, Mishra AK, Chattopadhyay DK, Raju KVSN (2007) Synthesis and characterization of hyperbranched polyesters and polyurethane coatings. J Polym Sci Part A Polym Chem 45:2673–2688
Poh AK, Sin LC, Foon CS, Hock CC (2014) Polyurethane wood adhesive from palm oil-based polyester polyol. J Adhes Sci Technol 28:1020–1033
Zhou J, Li H, Lu X (2018) Damping elastomer with broad temperature range based on irregular networks containing hyperbranched polyester and dangling chains. Polym Adv Technol 29:2308–2316
Manjula Dhevi D, Jaisankar SN, Pathak M (2013) Effect of new hyperbranched polyester of varying generations on toughening of epoxy resin through interpenetrating polymer networks using urethane linkages. Eur Polym J 49:3561–3572
Somisetti V, Allauddin S, Narayan R, Raju KVSN (2015) Synthesis of a novel glycerol based B3-type monomer and its application in hyperbranched polyester urethane–urea coatings. RSC Adv 5:74003–74011
Chan-Chan LH, Solis-Correa R, Vargas-Coronado RF, Cervantes-Uc JM, Cauich-Rodríguez JV, Quintana P, Bartolo-Pérez P (2010) Degradation studies on segmented polyurethanes prepared with HMDI, PCL and different chain extenders. Acta Biomater 6:2035–2044
Woodruff MA, Hutmacher DW (2010) The return of a forgotten polymer-Polycaprolactone in the 21st century. Prog Polym Sci 35:1217–1256
Kupka V, Vojtova L, Fohlerova Z, Jancar J (2016) Solvent free synthesis and structural evaluation of polyurethane films based on poly(ethylene glycol) and poly(caprolactone). Express Polym Lett 10:479–492
Oh JM, Lee SH, Son JS, Khang G, Kim CH, Chun HJ, Min BH, Kim JH, Kim MS (2009) Ring-opening polymerization of ε–caprolactone by poly(propyleneglcol) in the presence of monomer activator. Polymer 50:6019–6023
Zhao J, Xu R, Luo G, Wu J, Xia H (2016) Self-healing poly(siloxane-urethane) elastomers with remoldability, shape memory and biocompatibility. Polym Chem 7:7278–7286
Fukushima K, Feijoo JL, Yang M-C (2013) Comparison of abiotic and biotic degradation of PDLLA, PCL and partially miscible PDLLA/PCL blend. Eur Polym J 49:706–717
Gumede TP, Luyt AS, Müller AJ (2018) Review on PCL, PBS, and PCL/PBS blends containing carbon nanotubes. Express Polym Lett 12:505–529
Yuan S, Jiang S, Luo Y (2020) Cross-linking network structures and mechanical properties of novel HTPE/PCL binder for solid propellant. Polym Bull. https://doi.org/10.1007/s00289-020-03110-w
Unal S, Ozturk G, Sisson K, Long TE (2008) Poly(caprolactone) containing highly branched segmented poly(ester urethane)s via A2 with oligomeric B3 polymerization. J Polym Sci Part A Polym Chem 46:6285–6295
Džunuzović JV, Stefanović IS, Džunuzović ES, Dapčević A, Šešlija SI, Balanč BD, Lama GC (2019) Polyurethane networks based on polycaprolactone and hyperbranched polyester: structural, thermal and mechanical investigation. Prog Org Coat 13:105305
Vuković J (2006) Synthesis and characterization of aliphatic hyperbranched polyesters. PhD Dissertation, University of Osnabrück
Marand Å, Dahlin J, Karlsson D, Skarping G, Dalene M (2004) Determination of technical grade isocyanates used in the production of polyurethane plastics. J Environ Monit 6:606–614
Flory PJ, Rehner J (1943) Statistical mechanics of cross-linked polymer networks II. Swelling J Chem Phys 11:521–526
Brandrup J, Immergut EH (1989) Polymer Handbook, 3rd edn. John Wiley, New York
Hill LW (1997) Calculation of crosslink density in short chain networks. Prog Org Coat 31:235–243
Patil PN, Rath SK, Sharma SK, Sudarshan K, Maheshwari P, Patri M, Praveen S, Khandelwal P, Pujari PK (2013) Free volumes and structural relaxations in diglycidyl ether of bisphenol-A based epoxy–polyether amine networks. Soft Matter 9:3589–3599
Petrovic ZS, Ferguson J (1991) Polyurethane elastomers. J. Prog Polym Sci 16:695–836
Amrollahi M, Sadeghi GMM (2008) Evaluation of adhesion strength, flammability, and degradation of HBCD-containing polyurethane adhesives. J Appl Polym Sci 110:3538–3543
Bittiger H, Marchessault RH, Niegisch WD (1970) Crystal structure of poly-ε-caprolactone. Acta Crystallogr B 26:1923–1927
Woo HJ, Majid SR, Arof AK (2013) Effect of ethylene carbonate on proton conducting polymer electrolyte based on poly(ε-caprolactone) (PCL). Solid State Ion 252:102–108
Zhang J, Hu CP (2008) Synthesis, characterization and mechanical properties of polyester-based aliphatic polyurethane elastomers containing hyperbranched polyester segments. Eur Polym J 44:3708–3714
Džunuzović JV, Pergal MV, Poręba R, Vodnik VV, Simonović BR, Špírková M, Jovanović S (2012) Analysis of dynamic mechanical, thermal and surface properties of poly(urethane-ester-siloxane) networks based on hyperbranched polyester. J Non-Cryst Solids 358:3161–3169
Dash TK, Konkimalla VB (2012) Poly-є-caprolactone based formulations for drug delivery and tissue engineering: a review. J Control Release 158:15–33
Seyednejad H, Ghassemi AH, van Nostrum CF, Vermonden T, Hennink WE (2011) Functional aliphatic polyesters for biomedical and pharmaceutical applications. J Control Release 152:168–176
Gong CY, Fu SZ, Gu YC, Liu CB, Kan B, Deng HX, Luo F, Qian ZY (2009) Synthesis, characterization, and hydrolytic degradation of biodegradable poly(ether ester)-urethane copolymers based on e-caprolactone and poly(ethylene glycol). J Appl Polym Sci 113:1111–1119
Shamsi R, Sadeghi GMM (2016) Novel polyester diol obtained from PET waste and its application in the synthesis of polyurethane and carbon nanotube-based composites: swelling behavior and characteristic properties. RSC Adv 6:38399–38415
Schott H (1992) Swelling kinetics of polymers. J Pharm Sci 81(5):467–470
Emamikia M, Barikani M, Bakhshandeh G (2015) Relationship between structure and aromatic solvent permeability of crosslinked polyurethanes based on hyperbranched polyesters. Polym Int 64:1142–1154
Ritger PL, Peppas NA (1987) A simple equation for description of solute release. II Fickian and anomalous release from swellable devices. J Control Release 5:37–42
Alfrey TJ, Gurnee EF, Lloyd WG (1966) Diffusion in glassy polymers. J Polym Sci C 12:249–261
Fieldson GT, Barbari TA (1993) (1993) The use of FTi r-a t r spectroscopy to characterize penetrant diffusion in polymers. Polymer 34:1146–1153
Sarkar S, Adhikari B (2001) Synthesis and characterization of lignin-HTPB copolyurethane. Eur Polym J 37:1390–1401
Asif A, Shi W, Shen X, Nie K (2005) Physical and thermal properties of UV curable waterborne polyurethane dispersions incorporating hyperbranched aliphatic polyester of varying generation number. Polymer 46:11066–11078
Chattopadhyay DK, Webster DC (2009) Thermal stability and flame retardancy of polyurethanes. Prog Polym Sci 34:1068–1133
Zhang Y, Liao J, Fang XC, Bai F, Qiao K, Wang L (2017) Renewable highperformance polyurethane bioplastics derived from lignin-poly(epsilon-caprolactone). ACS Sustain Chem Eng 5:4276–4284
Cakić SM, Ristić IS, Krakovský I, Stojiljković DT, Bĕlský P, Kollová L (2014) Crystallization and thermal properties in waterborne polyurethane elastomers: Influence of mixed soft segment block. Mater Chem Phys 144:31–40
Kong X, Liu G, Curtis JM (2012) Novel polyurethane produced from canola oil based poly(ether ester) polyols: synthesis, characterization and properties. Eur Polym J 48:2097–2106
Wietor J-L, van Beek DJM, Peters GW, Mendes E, Sijbesma RP (2011) Effects of branching and crystallization on rheology of polycaprolactone supramolecular polymers with ureidopyrimidinone end groups. Macromolecules 44:1211–1219
Asif A, Huang CY, Shi WF (2005) Photopolymerization of waterborne polyurethane acrylate dispersions based on hyperbranched aliphatic polyester and properties of the cured films. Colloid Polym Sci 283:721–730
Xu Y, Chen D (2018) Shape memory-assisted self-healing polyurethane inspired by a suture technique. J Mater Sci 53:10582–10592
Stefanović IS, Dostanić J, Lončarević D, Vasiljević-Radović D, Ostojić S, Marković S, Pergal MV (2019) Preparation and characterization of poly(urethane-siloxane)/titanium-dioxide nanocomposites. Hem Ind 73:13–24
Fu S, Zhu J, Chen S (2018) Tunable shape memory polyurethane networks cross-linked by 1,3,5,7-tetrahydroxyadamantane. Macromol Res 26:1035–1041
Hazmi ASA, Pauzi NNPN, Maurad ZA, Abdullah LC, Aung MM, Ahmad A, Salleh MZ, Tajau R, Mahmood MH, Saniman SE (2017) Understanding intrinsic plasticizer in vegetable oil-based polyurethane elastomer as enhanced biomaterial. J Therm Anal Calorim 130:919–933
Acknowledgements
This work was financially supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (Grant No. 451-03-68/2020-14/200026). This research was financially supported in part by the Ministry of Education, Science and Technological Development of the Republic of Serbia through the former research projects: 172062 and III45007.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Stefanović, I.S., Džunuzović, J.V., Džunuzović, E.S. et al. Composition-property relationship of polyurethane networks based on polycaprolactone diol. Polym. Bull. 78, 7103–7128 (2021). https://doi.org/10.1007/s00289-020-03473-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-020-03473-0