
Abstract
In this study, to improve the thermo-mechanical properties of glycidyl azide polymer (GAP)-based elastomers, polyethylene adipate (PEA) with different molecular weights was successfully synthesized using a solvent-free method. GAP was synthesized by cationic ring-opening polymerization of epichlorohydrin and then azidation. The structure of PEA and GAP was characterized by FTIR, 1HNMR, and GPC. The compatibility of GAP/PEA at various weight ratio blends was investigated by differential scanning calorimetry. The result indicated GAP/PEA is compatible, and addition of PEA to GAP leads to the decrease in glass transition temperature. The copolyurethane elastomers were prepared by cross-linking GAP and PEA at various weight ratios using isophorone diisocyanate and N100 as curing agent. The thermo-mechanical properties were evaluated by dynamic mechanical analysis and tensile test. Based on the results, with increasing weight ratios of PEA, the copolyurethane elastomers displayed an increase in tensile strength (6.97 MPa) and elongation (792%) in comparison with GAP.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Solid composite propellants consist of oxidizers, metallic fuels, binders, plasticizer, and other additives [1,2,3]. Among these components, the binder plays an important role in binding the components of the propellant [4,5,6]. In the last decades, the use of polymeric binders including energetic functional groups in polymer backbones (such as nitro, azido, and difluoramine) [7, 8] instead of usual and inert binders such as hydroxyl-terminated polyether (HTPE), hydroxyl-terminated polybutadiene (HTPB), hydroxy‐terminated polyisoprene (HTPI), etc. [9, 10], to improve high-performance propellants is a novel field in energetic material [11,12,13]. Energetic polymeric binders improve the performance of the propellants by increasing the total energy of the formulations [14,15,16]. Among the energetic polymeric binders, azide polymers are considered in the solid composite propellant formulation. Glycidyl azide polymer (GAP) is a unique binder used in the propellant industry due to high density with positive heat of formation (+ 117.2 kcal/mol), a low tendency for detonation, and good compatibility with all high-energy oxidizers [17,18,19,20]. The usage of this binder in formulation leads to an increase in specific impulse, burning rate, decrease releases gas in combustion, and the best candidate polymer for chlorine-free eco-friendly solid composite propellant [21,22,23]. However, GAP is a well-known and promising energetic polymer with high polarity, rigidity, and conjugated structure of azide groups on the side chain as a pendent group in the polymer backbone, elastomers, or propellant based on GAP has low thermo-mechanical properties due to low flexibility and the existence methylene azido pending groups in its molecular structure [24,25,26]. Generally, the structural integrity of the propellant is affected by the polymeric binder characteristics. To overcome these problems, different methods have been used, for example, synthesis of higher molecular weight GAP, modification of the chain of GAP via interactions with other material, branched GAP, and blending of GAP with flexible chain polymers [27,28,29]. Blending two polymers is an easy method to improve the performance of solid propellants. This method has been developed in improving the mechanical properties of GAP propellant due to its efficiency and simplification. GAP which has high polarity azide groups grafted on the polymer backbone has good compatibility with energetic plasticizer, oxidizers, and other polar diols. Therefore, to modify the mechanical properties of elastomers based on GAP it can be used different polymers with flexible and polar structure backbones, such as polyethylene glycol (PEG), polycaprolactone (PCP) and poly(ethylene oxide-co-tetrahydrofuran) [P (EO-co-THF)] [30,31,32,33,34]. The mechanical properties of GAP-based propellant were improved by introduction of segmented GAP/PEG and GAP/PCL block copolymer networks to enhance the flexibility of polymer backbone [35]. Among polar and flexible polymer, polyester diol based on adipate has an excellent compatibility with polar polyols, high mechanical and thermal properties. Polyethylene adipate (PEA) is an aliphatic crystalline polyester synthesized from monoethylene glycol and adipic acid, which presents the nature of biodegradability [36,37,38].
In this study, to improve the thermo-mechanical properties of GAP, the blending of GAP/PEA is considered. We investigate the compatibility of these blends with differential scanning calorimetry (DSC). The copolyurethane network elastomers are prepared using GAP and different contents of PEA as a diol and isophorone diisocyanate (IPDI)/desmodure N100 as curing agents. The thermo-mechanical properties were evaluated by dynamic mechanical analysis (DMA) and tensile test.
Experimental
Materials and methods
Epichlorohydrin (ECH) 99%, 1,4-butanediol (BDO) 99%, boron trifluoride etherate (BF3.OEt2), dichloromethane (DCM) > 99.8, sodium azide 99.5%, dimethylformamide (DMF) ≥ 99.8%, magnesium sulfate (MgSO4) ≥ 99.5%, mono ethylene glycol (MEG) ≥ 99.0%, adipic acid (AA) ≥ 99.0%, isophorone diisocyanate (IPDI) 99%, titanium isopropoxide (TIP) 97%, and dibutyltin dilaurate (DBTDL) ≥ 96.0% were purchased from Merck Company. Desmodur N100 (N100) with NCO content (%NCO = 22) was purchased from Bayer. All solvents dried in vacuum at 60 °C for 24 h before use.
The FTIR spectra of the materials were recorded on a Bruker FTIR spectrometer (KBr pellet) in the spectral range of 400–4000 cm−1. The ATR-FTIR spectrum was carried out by the GOLDEN GATE model from company SPECAC. Zinc selenide (ZnSe) prism is used in the ATR accessory. 1H NMR spectra are recorded on a Bruker spectrometer on 500 MHz at ambient temperature with CDCl3 as solvent. TMS or tetramethylsilane is used as an internal standard for calibrating chemical shift in 1H NMR. NCO content of IPDI and N100 was determined using ASTM D2572. The hydroxyl value (OHV) indicates the necessary amount of KOH (in mg) to neutralize the consummated amount of acetic acid of 1 g fat during acetylation and was determined according to ASTM D4247. The hydroxyl values (mg KOH/g) were calculated by the following equation:
where A, B, N, and W are the volume (ml) of potassium hydroxide solution used for the titration of the blank (without sample), volume (ml) of potassium hydroxide solution used for the titration of a sample, normality of potassium hydroxide solution, and the weight (g) of sample, respectively.
The acid numbers of the polyester diols were determined by ASTM D1980. Gel permeation chromatography (GPC) was performed using a GPC Agilent 1100 (USA) instrument with a refractive index detector, using an Agilent PLgel 5 µm mixed-C 300 × 7.5 mm column; THF was used as the solvent and injected at 30 °C at the rate of 1 mL/min and calibrated with polystyrene standard. Tensile tests were performed at the Hiwa 200 instrument with a crosshead speed of 50 mm/min at room temperature and 50 ± 5% relative humidity. All gauge sizes and shapes were compatible with the standard D638-02a (type IV). A PerkinElmer STA 6000 instrument differential scanning calorimeter was used to determine the glass transition temperature (Tg) under a nitrogen atmosphere at a heating rate of 20 °C/min. The dynamical mechanical thermal analysis (DMTA) test was carried out using a Perkin 800 instrument with a film tension mode at 1 Hz. This analysis was performed in a nitrogen atmosphere, at a heating rate of 5 °C min−1, in the temperature range of − 100–100 °C.
Synthesis of glycidyl azide polymer
The glycidyl azide polymer was synthesized in two stages. The first stage was the synthesis of polyepichlorohydrine by cationic ring-opening polymerization. The second stage was the synthesis of GAP from polyepichlorohydrine by azidation. The preparation of GAP was carried out in a three-necked round bottom flask connected to a nitrogen inlet, thermometer, and magnetic bar. In this reaction, BDO (11 g, 0.122 mol) was dissolved in 400 ml distilled DCM. Then, BF3.OEt2 (5 ml, 0.040 mol) was added into the reaction mixture and stirred at room temperature for 30 min. Epichlorohydrin (340 g, 3.675 mol) was added dropwise to the reaction flask during 12 h. The reaction mixture was stirred overnight at room temperature under nitrogen atmosphere. Finally, the reaction solution was quenched by adding 50 g sodium bicarbonate solution. The organic layer was washed with distilled water and then dried with magnesium sulfate and filtered. The solvent and unreacted monomers were removed by vacuum evaporation. Polyepichlorohydrine was obtained after dried under vacuum at 30 °C (325 g, Yield: 92%). The molar ratio of initiator to monomer (MW = 92.5 g mol−1) was 1:30 until molecular weight obtained in rang of 2500–3000 g mol−1.
In the second step, in a three-necked flask connected to the thermometer, condenser, and magnetic bar polyepichlorohydrine (300 g) was dissolved in DMF (600 ml). Then sodium azide (240 g, 3.7 mol) was slowly added to the mentioned solution at 60 °C for 30 min. The reaction was heated to 100 °C and continues under stirring for 15 h and then was cooled, and solid materials were filtered. The organic phase was separated and washed with distilled water, dried with magnesium sulfate, and filtered. The solvent was removed by vacuum evaporation to obtain the yellow GAP (270 g, Yield: 90%).
Synthesis of polyethylene adipate
The polyester diols, PEA (Mn = 1000 g/mol), were synthesized by polycondensation reaction of AA and MEG in the presence of Ti (i-Pr)4 as a catalyst [38]. The reaction was carried out in a three-necked round bottom flask equipped with a dean & Stark trap, thermometer, condenser, and mechanical stirrer. The flask was charged with MEG (43.4 g, 0.7 mol), AA (87.6 g, 0.6 mol), and Ti(i-Pr)4 heated to 130 °C to achieve a clear liquid. Then, by collection of water as a byproduct in the Dean and Stark trap at 190 °C, the PEA synthesis was monitored. Finally, the acid value was determined by titration and reached 3. After slowly reduction of pressure to below 100 mm Hg and increase in temperature to 220 °C, water was collected and acid value reached to 1 mg KOH/g. The reaction mixture was cooled down to room temperature, and white solid product was obtained (109 g, yield: 83.5%). These methods were used for varying the molecular weight.
Polymer blend preparation
GAP and PEA have dried in vacuum (200 mmHg) at 70 °C for 24 h before blending. Polymer blends were prepared through balk methods by mixing. The blending of GAP and PEA with different weight compositions (90/10, 80/20, 70/30, 60/40, and 50/50) was carried out in 60 °C for 60 min. Then, these compositions were stored at room temperature for three days to investigate any phase separation or multi-layer formation.
Preparation of polyurethane networks
To prepare the GAP/PEA-based polyurethane networks elastomer, GAP was placed in a three-necked flask fitted with a thermometer and nitrogen inlet, in an oil bath and heated to 60 °C under vacuum for 2 h. PEA, IPDI, and N100 at a NCO/OH ratio of 1:1 were added to flask and stirred for 30 min; then, DBTDL (0.1 wt%) was added to mixture as catalyst. The solution was stirred for 20 min under vacuum pump for degassing at 50 °C. The mixture was poured into teflon-coated mold and left to oven for 7 days at 70 °C. A preparation method of copolyurethane network elastomers is summarized in Scheme 1.

Process of preparation of copolyurethane binder
Results and discussion
Structures and characterization of GAP
GAP was synthesized by cationic ring-opening polymerization of ECH, using BDO as an initiator and BF3.OEt2 as a catalyst, followed by azidation with sodium azide in DMF. Scheme 2 shows the pathway for the synthesis of GAP. The structure of GAP was characterized by FT-IR and 1H NMR spectra.

Synthesis route of GAP
The FT-IR spectra (Fig. 1) show successfully synthesized of polyepichlorohydrin and GAP. As shown in Fig. 1a, the broad absorption band at 3300–3550 cm−1 is related to the hydroxyl end groups. The peaks around 2924 cm−1 and 2874 cm−1 are assigned to C–H symmetric and antisymmetric stretching vibration of polyepichlorohydrin. The characteristic peaks at 1128 cm−1 and 750 cm−1 attributed to symmetric stretching vibrations of C–O and C–Cl group. In the FTIR spectrum of GAP (Fig. 1b), the absorption band related to C–Cl disappears at 750 cm−1 and the characteristic peak of − N3 appears at 2102 cm−1. All these peaks confirm the successful synthesize of GAP.

FT-IR spectra of polyepichlorohydrin (A) and GAP (B)
The structures of polyepichlorohydrin and GAP were also characterized by 1H NMR spectra as presented in Fig. 2. As shown in Fig. 2a, characteristic peak observed at 1.5–1.7 ppm attributed to –CH2–CH2—of the 1,4-butanediol (denoted b), 3.53–3.7 ppm of –CH2–Cl protons of pendent group (denoted d), 3.75–3.85 ppm related to methyne and methylene protons in polyether backbone of polyepichlorohydrin (denoted c, e), and small peak at 3.9–4 ppm related to terminal hydroxyl group (denoted a). As shown in Fig. 2b, after azidation of polyepichlorohydrin, the peak at 3.53–3.7 ppm related to the methylene protons of chloromethyl groups (–CH2–Cl) disappeared and a new peak observed at 3.2–3.5 ppm related to the resonance of the methylene protons (denoted d) of azidomethyl groups. Therefore, as a result of FT-IR and 1H NMR, chlorine atoms were successfully replaced by azide groups. Also, the molecular weight of GAP was calculated by 1H NMR and was obtained 2570 g/mol. According to the 1H NMR spectrum of GAP, the peaks of protons (b) and (d) attributed to methylene protons of the 1,4-butanediol and azidomethyl groups, respectively, appear clearly and do not overlap with any other peaks. Therefore, these two peaks were used to calculate molecular weight. From the integral ratio of protons (b) and (d) in 1H NMR and the ratio of the number of the protons in the theoritical GAP structure, the molecular weight of GAP can be calculated.

1HNMR spectra of polyepichlorohydrin (A) and GAP (B)
Structures and characterization of PEA
The polyethylene adipate (PEA) were synthesized via polycondensation reaction of MEG and AA under vacuum and high temperature. The synthesis route of PEA is illustrated in Scheme 3.

Synthesis route of PEA
In this reaction, water is collected as a byproduct to reach appropriate acid value (AN < 2). The chemical and chain structures of the PEA were characterized by FT-IR (Fig. 3) and 1H NMR (Fig. 4) spectra. For the PEA diol, the sharp stretching vibrations of the C = O ester group appeared at 1756 cm−1, and the band appeared at 1250 cm−1 related to the (C = O)–O groups. The bands ascribed to stretching vibrations of the CH2 groups were observed at 2870 to 2920 cm−1. The wide absorbed band at 3200–3600 cm−1 is related to terminal OH groups.

FT-IR spectra of PEA

1HNMR spectra of PEA
Also, the structure of PEA was confirmed by 1H NMR spectroscopy. The 1H NMR spectra of PEA show the single at δ:4–4.2 ppm attributed to the protons of the ester group (denoted b), triplet signal at 3.6 ppm related to –CH2OH terminal groups (denoted a), and triplet signals at 2.5 ppm and 1.5 ppm attributed to –CH2—of adipate segments denoted c and d, respectively. According to the 1H NMR spectrum of PEA, the peaks of protons (a) and (b) always appear clearly and do not overlap with any other peaks. Therefore, these two peaks were used to calculate molecular weight. From the integral ratio of protons (a) and (b) in 1H NMR and the ratio of the number of the protons in the theorical PEA structure the molecular weight of PEA can be calculated. The characterization of synthesized materials is summarized in Table 1. As can be seen, the calculated molecular weight by the three methods is different. Since GPC is a relative method, determined molecular weight of the samples is relative values and they are described as polystyrene equivalent molecular weight. In the 1H NMR spectrum of PEA, integral ratios of the resonance signals in every spectrum are equal to their ratios of these protons in the polymer structure and the hydroxyl number represents the hydroxyl content of the material. In comparison with gel permeation chromatography (GPC), 1H NMR and hydroxyl number have the considerable advantages to calculate molecular weight along exact results. The calculated molecular weight by hydroxyl number is used for preparation of these polyurethanes.
Compatibility study on GAP/PEA blends
The glass transition temperatures (Tg) of polymers compositions are useful method for compatibility study of polymer systems. The obtained single-point Tg values for polymers compositions are an indication of homogeneity, which confirms the thermodynamic compatibility of polymers compositions. GAP is a polar polymer with polyether backbone and azide pendent groups; also PEA is a polar polymer with flexible ester group in the backbone. Hence, GAP and PEA be miscible, and phase separation will not occur for the blends. First, different ratios of GAP/PEA (90:10, 70:30, and 50:50) were mixed in test tube. Three days after the preparation of the resultant compositions at 60 °C, no sign of heterogeneity was observed in all of the samples, denoting the physical compatibility of the GAP with the PEA. The DSC curves of the GAP/PEA blends from different weight ratios are shown in Fig. 5, and their Tg values are summarized in Table 2. According to results, with the increase in weight ratio of PEA content only one Tg is observed and decreases from − 45.2 to − 47.1 °C. The obtained single point Tg values confirms the thermodynamic compatibility of all GAP /PEA compositions.

Glass transition temperature of GAP/PEA blends
Structures and characterization of copolyurethane networks
Polyurethane network structures are prepared via the reaction of GAP and PEA with the mixed isocyanate (IPDI/N100) curing agent IPDI and N100. A schematic of this reaction is shown in Scheme 4. The structures of copolyurethane networks were characterized by ATR spectroscopy.

Synthesis route of copolyurethane elastomer based on GAP/PEA
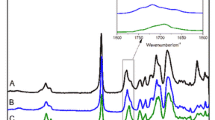
The ATR spectrum of PU film is shown in Fig. 6. As can be seen, the broad absorption band related to hydroxyl of polyol and the characteristic peak of isocyanate groups disappears at 3400 cm−1 and 2270 cm−1, respectively. The characteristic peak of urethane appears at 1730 cm−1, indicating isocyanato and hydroxyl in the system react.

ATR spectra of copolyurethane elastomer based on GAP/PEA
Tensile properties of copolyurethane networks
First, the effect of three different molecular weights of PEA on the tensile properties at 25 °C was studied. As shown in Fig. 7, the PU network elastomers derived from PEA-3000 and PEA-2000 exhibited the stress–strain behavior of plastic deformation, while PU network elastomers based on PEA-1000 exhibited the elastic behavior and yield point of usual elastomeric polymers. According to results, the tensile strength and Young’s modulus of the PU network elastomers are increasing with the increase in weight ratio of PEA content; meanwhile, crystalline behavior in PU network elastomers is increased. Therefore, PU network elastomers based on PEA-3000 indicate higher mechanical properties compared to PEA-1000 and PEA-2000.

Stress–strain curves of PEA-based networks
The results of the mechanical properties of PU network elastomers based on PEA are summarized in Table 3. According to results, PEA-3000 was selected for blending with the GAP due to excellent mechanical properties.
The tensile curves of copolyurethane network elastomers from different weight ratios of GAP/PEA-3000 blends are shown in Fig. 8. The addition of neutral diol such as PEA to propellant systems leads to decreasing in energy performance. The GAP/PEA networks were prepared from PEA whose content varied from 0 to 50 wt%. As shown in Fig. 8, mechanical properties of copolyurethane network elastomers based on pure GAP were poor as tensile strength, and elongation at break was 0.3 MPa and 178.2%, respectively. The results clearly point out that “addition of PEA” is highly effective in improving the mechanical properties. In the GAP/PEA samples containing 10 wt% and 20 wt% of PEA, improving mechanical properties are less obvious, while PEA concentrations higher than 20 wt% noticeably improved the mechanical properties of elastomer. For these elastomers, tensile strength values and elongation at break increased significantly. Thus, the addition of PEA to GAP has a large effect on mechanical properties of elastomer such as Young’s modulus, tensile strength, and elongation at break. The results are summarized in Table 4.

Stress–strain curves of copolyurethane networks with various contents of PEA
Thermo-mechanical properties of copolyurethane networks
The thermal and mechanical properties of the copolyurethane network were investigated using DMTA. Figures 9 and 10 show the storage modulus and tan δ versus temperature ranging from − 80 to 100 °C for copolyurethane network from different weight ratios of PEA-3000 in GAP. The glass transition temperature (Tg, α relaxation) for copolyurethane networks was determined from tan δmax (Table 5). At low temperatures, all copolyurethane networks were in the glassy state with E′ values on the order of 2000 MPa. As temperature increased, E′ of copolyurethane networks gradually decreased, and near glass transition temperature, a rapid decrease was observed. At higher temperatures, the storage modulus of all copolyurethane networks reached a rubbery plateau. All samples show only one tan δ peak, indicating the homogeneous nature of the copolyurethane network elastomers. The second peak in PEA content 40% and 50% in 32.5 °C is related to the melting point of the crystalline segment of PEA. As the PEA content increased in copolyurethane network from 10 to 50%, the tan δ peak of the respective copolyurethane network elastomers shifted from 3.1 to − 28.1 °C. This is attributed to the fact that increasing of PEA content caused increasing soft and flexible segment, and thus, glass transition temperature decreased.

Storage modulus versus temperature curves of copolyurethane networks

Tan δ versus temperature curves of copolyurethane networks
The addition of PEA to GAP in the presence of IPDI/N100 leads to the formation of interpenetrating polymer networks and enhances both the chemical crosslinking density and the physical interaction such as hydrogen bonding between chains in the copolyurethane network elastomers, which was improved the mechanical properties. Also, addition of PEA to GAP causes a decrease in bulk azide pending groups in GAP; therefore, molecular chain can move more easily that decreases Tg values and leading to increasing mechanical properties of copolyurethane network elastomers such as Young’s modulus, tensile strength, and elongation at break than pure GAP network elastomer.
The crystallization of the PEA segments causes the high physical crosslinking density in copolyurethane networks elastomers including more than 40 wt % PEA. The effect of PEA in the copolyurethane network elastomers is shown in Scheme 5.

Description of copolyurethane network elastomer: a GAP-based polyurethane network elastomer; b copolyurethane network elastomer of GAP/PEA (PEA < 40 wt%); c copolyurethane network elastomer of GAP/PEA (PEA > 40 wt %)
Conclusions
The GAP/PEA blend with various weight ratios of 90/10, 80/20, 70/30, 60/40, and 50/50 was prepared and compatibility of these blends investigated with DSC. The single-point Tg values were confirmed compatibility of this two kind diols. Also addition of PEA to GAP leads to decrease in Tg from − 44.7 to − 47.1 °C for GAP/PEA = 50/50 weight ratio. The tensile test demonstrates that PEA-3000 has better mechanical properties compared to the PEA-1000 and PEA-2000. The copolyurethane network elastomers were prepared with different contents of PEA-3000 (content varied from 0 to 50 wt%.) and characterized with ATR, DMA, and tensile test. According to analysis, increasing the PEA content of the copolyurethane network elastomers resulted in a decrease in Tg values and leading to increasing mechanical properties of copolyurethane network elastomers such as Young’s modulus, tensile strength, and elongation at break than pure GAP network elastomer. Thus, the addition of PEA to GAP-based elastomers, especially 50% of PEA, indicated superior tensile properties (tensile strength = 6.97 MPa and elongation at break = 792%) due to the crystalline segment in elastomers were increased.
References
Deng J, Li G, Xia M, Lan Y, Luo Y (2016) Improvement of mechanical characteristics of glycidyl azide polymer binder system by addition of flexible polyether. J Appl Polym Sci 133(35):43840. https://doi.org/10.1002/app.43840
Azoug A, Constantinescu A, Nevière R, Jacob G (2015) Microstructure and deformation mechanisms of a solid propellant using 1H NMR spectroscopy. Fuel 148:39–47
Zhang Z, Luo N, Wang Z, Luo Y (2015) Polyglycidyl nitrate (PGN)-based energetic thermoplastic polyurethane elastomers with bonding functions. J Appl Polym Sci. https://doi.org/10.1002/app.42026
Min BS, Ko SW (2007) Characterization of segmented block copolyurethane network based on glycidyl azide polymer and polycaprolactone. Macromol Res 15(3):225–233
Murali Mohan Y, Padmanabha Raju M, Mohana Raju K (2004) Synthesis, spectral and DSC analysis of glycidyl azide polymers containing different initiating diol units. J Appl Polym Sci 93(5):2157–2163. https://doi.org/10.1002/app.20682
Khanlari T, Bayat Y, Bayat M (2019) Synthesis, thermal stability and kinetic decomposition of triblock copolymer polypropylene glycol–poly glycidyl nitrate–polypropylene glycol (PPG–PGN–PPG). Polym Bull. https://doi.org/10.1007/s00289-019-03051-z
Bhowmik D, Sadavarte VS, Pande SM, Saraswat BS (2015) An energetic binder for the formulation of advanced solid rocket propellants. Cent Eur J Energ Mat 12(1):145–158
Xu M, Ge Z, Lu X, Mo H, Ji Y, Hu H (2017) Fluorinated glycidyl azide polymers as potential energetic binders. RSC Adv 7(75):47271–47278
Badgujar D, Talawar M, Asthana S, Mahulikar P (2008) Advances in science and technology of modern energetic materials: an overview. J Hazard Mater 151(2–3):289–305. https://doi.org/10.1016/j.jhazmat.2007.10.039
Iwama A, Hasue K, Takahashi T, Matsui K, Ishiura K (1996) Hydrogenated Hydroxy-Terminated Polyisoprene as a Fuel Binder for composite solid propellants. Propellants, Explos, Pyrotech 21(1):43–50. https://doi.org/10.1002/prep.19960210110
Badgujar D, Talawar M, Asthana S, Mahulikar P (2008) Advances in science and technology of modern energetic materials: an overview. J Hazard Mater 151(2–3):289–305. https://doi.org/10.1016/j.jhazmat.2007.10.039
Gaur B, Lochab B, Choudhary V, Varma I (2003) Azido polymers energetic binders for solid rocket propellants. J Macromol Sci Polymer Rev 43(4):505–545
Simić D, Petković J, Milojković A, Brzić S (2013) Influence of composition on the processability of thermobaric explosives. Sci Tech Rev 63(3):3–8
Klapötke TM, Sproll SM (2010) Investigation of nitrogen-rich energetic polymers based on alkylbridged bis-(1-methyl-tetrazolylhydrazines). J Polym Sci Polym Chem 48(1):122–127. https://doi.org/10.1002/pola.23767
Schöyer H, Schnorhk A, Korting P, Va P, Mul J, Gadiot G, Meulenbrugge J (1995) High-performance propellants based on hydrazinium nitroformate. J Propul Power 11(4):856–869. https://doi.org/10.2514/3.23911
Liu D, Geng D, Yang K, Lu J, Chan SHY, Chen C, Hng HH, Chen L (2020) Decomposition and energy-enhancement mechanism of the energetic binder glycidyl azide polymer at explosive detonation temperatures. J Phys Chem. https://doi.org/10.1021/acs.jpca.0c02950
Hagen TH, Jensen TL, Unneberg E, Stenstrøm YH, Kristensen TE (2015) Curing of Glycidyl Azide Polymer (Gap) Diol Using Isocyanate, Isocyanate-Free, Synchronous Dual, and Sequential Dual Curing Systems. Propellants Explos Pyrotech 40(2):275–284. https://doi.org/10.1002/prep.201400146
Zhao Y, Zhang X, Zhang W, Xu H, Xie W, Du J, Liu Y (2016) Simulation and experimental on the solvation interaction between the GAP matrix and insensitive energetic plasticizers in solid propellants. J Phys Chemi 120(5):765–770. https://doi.org/10.1021/acs.jpca.5b10540
Tanver A, Rehman F, Wazir A, Khalid S, Ma S, Li X, Luo Y, Huang M-H (2016) Energetic hybrid polymer network (EHPN) through facile sequential polyurethane curation based on the reactivity differences between glycidyl azide polymer and hydroxyl terminated polybutadiene. RSC Adv 6(13):11032–11039. https://doi.org/10.1039/C5RA23250C
Li Y, Li J, Ma S, Luo Y (2017) Compatibility, mechanical and thermal properties of GAP/P (EO-co-THF) blends obtained upon a urethane-curing reaction. Polym Bull 74(11):4607–4618
Manu S, Varghese T, Mathew S, Ninan K (2009a) Studies on structure property correlation of cross-linked glycidyl azide polymer. J Appl Polym Sci 114(6):3360–3368
Ma S, Li Y, Li G, Luo Y (2017) Research on the mechanical properties and curing networks of energetic GAP/TDI binders. Cent Eur J Energ Mat 14(3):708–725
He L, Zhou J, Wang Y, Ma Z, Chen C (2020) Mechanical and thermal properties of polyether polytriazole elastomers formed by click-chemical reaction curing glycidyl azide polymer. Molecules 25(8):1988. https://doi.org/10.3390/molecules25081988
Mohan YM, Raju KM (2005) Synthesis and characterization of HTPB-GAP cross-linked co-polymers. Des Monomers Polym 8(2):159–175. https://doi.org/10.1163/1568555053603215
Stacer RG, Husband DM (1991) Molecular structure of the ideal solid propellant binder. Propellants Explos Pyrotech 16(4):167–176. https://doi.org/10.1002/prep.19910160404
Bui V, Ahad E, Rheaume D, Raymond M (1996) Energetic polyurethanes from branched glycidyl azide polymer and copolymer. J Appl Polym Sci 62(1):27–32. https://doi.org/10.1002/(SICI)1097-4628(19961003)62:1%3c27::AID-APP5%3e3.0.CO;2-U
Mohan YM, Raju KM (2006) Synthesis and characterization of GAP-THF copolymers. Int J Polym Mater 55(3):203–217. https://doi.org/10.1080/009140390925134
Min BS, Baek G, Ko SW (2007) Characterization of polyether-type GAP and PEG blend matrices prepared with varying ratios of different curatives. J Ind Eng Chem 13(3):373–379
Manu S, Varghese T, Mathew S, Ninan K (2009b) Compatibility of glycidyl azide polymer with hydroxylterminated polybutadiene and plasticizers. J Propul Power 25(2):533–536. https://doi.org/10.2514/1.38145
Mathew S, SeK M, TeL V (2008) Thermomechanical and morphological characteristics of cross-linked GAP and GAP–HTPB networks with different diisocyanates. Propellants Explos Pyrotech 33(2):146–152. https://doi.org/10.1002/prep.200800213
Davenas A (2003) Development of modern solid propellants. J Propul Power 19(6):1108–1128. https://doi.org/10.2514/2.6947
DeLuca L, Galfetti L, Maggi F, Colombo G, Merotto L, Boiocchi M, Paravan C, Reina A, Tadini P, Fanton L (2013) Characterization of HTPB-based solid fuel formulations: Performance, mechanical properties, and pollution. Acta Astronaut 92(2):150–162. https://doi.org/10.1016/j.actaastro.2012.05.002
Mohan YM, Raju KM (2004) Synthesis and characterization of low molecular weight azido polymers as high energetic plasticizers. Int J Polym Anal Ch 9(5–6):289–304. https://doi.org/10.1080/10236660490935709
Sun Min B (2008) Characterization of the plasticized GAP/PEG and GAP/PCL block copolyurethane binder matrices and its propellants. Propellants Explos Pyrotech 33(2):131–138. https://doi.org/10.1002/prep.200700241
Teramoto N, Saitoh Y, Takahashi A, Shibata M (2010) Biodegradable polyurethane elastomers prepared from isocyanate-terminated poly (ethylene adipate), castor oil, and glycerol. J Appl Polym Sci 115(6):3199–3204. https://doi.org/10.1002/app.30019
He X, Qiu Z (2018) Influence of high molecular weight poly(ethylene adipate) on the crystallization behavior and mechanical properties of biodegradable poly (l-lactide) in their immiscible polymer blend. Polym Test 67:421–427. https://doi.org/10.1016/j.polymertesting.2018.03.038
Kim M-N, Kim K-H, Jin H-J, Park J-K, Yoon J-S (2001) Biodegradability of ethyl and n-octyl branched poly (ethylene adipate) and poly (butylene succinate). Euro Polym J 37(9):1843–1847. https://doi.org/10.1016/S0014-3057(01)00003-9
Shendi HK, Omrani I, Ahmadi A, Farhadian A, Babanejad N, Nabid MR (2017) Synthesis and characterization of a novel internal emulsifier derived from sunflower oil for the preparation of waterborne polyurethane and their application in coatings. Prog Org Coat 105:303–309. https://doi.org/10.1016/j.porgcoat.2016.11.033
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bayat, Y., Kashef Shandi, H. & Khanlari, T. Superior improvement in thermo-mechanical properties of polyurethane based on glycidyl azide polymer/polyethylene adipate. Polym. Bull. 78, 6577–6593 (2021). https://doi.org/10.1007/s00289-020-03440-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-020-03440-9