
Abstract
Novel core–shell nanofiber structures loaded by an essential oil using chitosan (CH) as a polymer have been successfully produced via the simple and effective production method of coaxial electrospinning. For this purpose, origanum minutiflorum (OM) oil was incorporated into the nanofibers. A blended form of the nanofibers (B–OM) was obtained by simply mixing OM with CH polymer solution, then this blended form was loaded separately into the core (C–OM) and the shell (S–OM) layers to obtain different composite core–shell nanofiber structures. The structures of the core and shell layers were investigated by TEM analysis. Furthermore, water contact angle analysis confirmed composition of the shell layer of each nanofiber type of B–OM, S–OM, C–OM, and differentiated it from the monolithic nanofiber of CH. The SEM images displayed the average diameter of the C–OM as 291 ± 10, while S–OM nanofibers demonstrated 284 ± 12 nm. The S–OM composite nanofibers showed the highest antibacterial activity during 24 h of the testing time. The nanofiber mats of B–OM and S–OM showed initial burst release with different profiles over an extended 7-day period of time after investigation with an in vitro drug release test. Moreover, C–OM nanofibers demonstrated prolonged time for in vitro drug release behavior with the initial burst profile at 8 h, then the release profile was relatively slow and sustained for about 7 days. The OM oil included nanofiber mats with different core–shell and blended morphologies that can hold a great promise for wound healing, antibacterial, and biomedical applications due to the controlled and tunable drug release and antibacterial activities. Another important advantage of our method over the traditional techniques is being eco-friendly, since it uses natural compound and natural polymer with controllable gas permeability of the nanofiber porous structure.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Electrospinning is one of the popular methods to produce various and controlled structures and morphologies of nanofibers using a wide range of polymers [1,2,3]. Electrospun nanofibrous mats are featured by controllable pore volumes, large surface area to mass ratio with a low density to obtain a very thin layer of nonwovens which can be designed for next-generation nanofiber-based wound dressings and many other advanced structures for different applications [4, 5]. Nanofibers can be engineered in a controllable fashion to demonstrate the unique capabilities of architectures such as core–shell [6], hollow [7], and porous [8] structures that are some of the important types. Core–shell nanofibers can be produced via a simple method of coaxial electrospinning [2]. They have become progressively important in different fields by demonstrating better performance in drug loading and displaying release behavior over a long period of time [9, 10]. Numerous drugs and bioactive agents such as growth factors, proteins, DNA, and antibiotics can be directly incorporated into the core and protected by the shell polymeric layer [11].
A controlled drug release system is difficult to achieve with the conventional methods, so some novel approaches have been employed to enhance the drug release and therapy behavior [12]. Some commonly featured reasons for applying the controlled drug delivery systems can be outlined as increasing patient’s compliance, decreasing premature drug release, helping drugs to cross physiological barriers, and superior control of the release that happens over a long time [13].
Wound dressings are a great way to speed up the wound healing process with its minimally invasive and effective manner. Recently, in addition to the traditional protecting materials, the core–shell nanofiber-based multifunctional dressings have become available to promote the healing performance by providing antimicrobial properties. As a result, various types of modern wound dressings have been evolved from honey pastes and natural plant fibers and made into the gauzes, bandages and artificial cloths and tissues led by the development of technology [14,15,16].
On the other hand, the plant extracts play an important role in the wound healing process because they have a detrimental effect on the pathogenic bacteria and show persistent protection along with the healing until the proliferation phase of the wound is completed [17]. In this investigation, we utilized the plant, Origanum Minutiflorum. This plant has been known as wild oregano since ancient times in food seasoning [18]. Origanum Minutiflorum as a natural oil plant has good potential to provide a suitable environment for strong antibacterial activity [19]. According to the literature, the antibacterial properties of the essential oils can be followed as: oregano > clove > coriander, and so on [18]. Therefore, OM-based dressing has very strong potential for the wound healing process.
In addition, chitosan is a typically famous biomaterial which has antifungal and antibacterial properties in addition to demonstrating biodegradability, biocompatibility and wound healing behavior [20]. Many studies have proved that the chitosan-based dressings can facilitate the contraction of wounds, accelerate the repair of different tissues, and regulate secretion of inflammatory mediators, such as prostaglandin E, interleukin 1β, and interleukin 8 [21].
In the present study, to control the antibacterial activity and drug release profile, CH-based core–shell nanofibers in which OM loaded into the core and shell parts separately by coaxial spinneret were obtained for the first time. The morphology and drug release behavior of these nanofibers were investigated and compared. The presence of OM in the nanofibers was determined using FTIR, TEM, and water contact angle measurement. Moreover, the drug release behavior was studied through UV–Vis spectrophotometer. Antibacterial activity was evaluated against gram-positive bacteria of Staphylococcus aureus and gram-negative bacteria of Pseudomonas aeruginosa. It is worth to note that the release profile and antibacterial properties can be controlled by obtaining different structures of nanofibers, although the same drug and polymer were used. These nanofeatured constructs can provide numerous implications in a broad range of biomedical applications where either burst or sustained drug release is important.
Experimental
Materials
A high molecular weight CH polymer of 310 kg/mol with > 75% degree of acetylation was obtained from Sigma-Aldrich. The extraction of OM oil by hydro-distillation method was purchased from a local market in Turkey. To solve CH polymer, trifluoroacetic acid (TFA) and dichloromethane (DCM) were supplied from Merck Millipore and Carlo Erba Reagents S.A.S, respectively. Water for chromatography was purchased from Merck KGaA to measure the water contact angle. Unless otherwise specified, all the chemicals were used as received without further purification.
Solution preparation
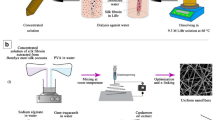
CH polymer solution (8% w/v) was prepared by dissolving it in TFA:DCM (3:2 v/v) with continuous stirring at a speed of 700 rpm for 4 h at 70 °C to obtain a homogeneous mixture. Then, the drug content was adjusted to 10 wt% in CH solution by adding OM extract as seen in Fig. 1. Both of the solutions were used to obtain different nanofiber structures as shown in Table 1. Parts 2.3 and 2.4 explain the preparation of the nanofiber mats using the single and coaxial nozzles.

A photograph of CH, OM and B–OM solutions
Single nozzle electrospinning process
Pure CH and B–OM solutions were used separately for electrospinning to obtain virgin and composite nanofibers. The nanofibers were electrospun using the basic electrospinning unit (NanoWEB-101, Turkey) and collected on an aluminum plate covered by aluminum foil. The voltage, tip to the collector distance and flow rate of electrospinning unit were adjusted with 25 kV, 15 cm and 2 mlh−1, respectively. A syringe with the volume of 2.5 ml was placed horizontally on a programmable single syringe pump during the electrospinning process.
Coaxial electrospinning process
Two different morphologies of core–shell structures were electrospun. In the first sample, CH polymer was fed to the core part and B–OM was incorporated into the shell part to produce S–OM nanofiber mats. The coaxial electrospinning was performed using coaxial spinneret with 26 kV voltage, 15 cm distance and then flow rate of core and shell were fixed at 0.2 and 0.7 mlh−1, respectively. In the second sample, B–OM and CH were used for the core and shell parts, respectively, to spun C–OM nanofiber mats. Voltage and distance of the core and the shell parts were adjusted at 26 kV and 15 cm with the flow rate of 0.2 and 1 mlh−1, respectively. All the experiments were performed under room temperature.
Characterization
Several complementary analytical techniques were employed to characterize the electrospun nanofibers including scanning electron microscope, transmission electron microscopy, X-ray diffraction, differential scanning calorimetry, and fourier transform infrared spectroscopy. All of these analyses are explained in detail as follows:
Scanning electron microscope
The size, shape and morphology of the electrospun nanofibers were observed with the scanning electron microscope (SEM, JEOL JSM-5600LV microscope). The accelerating voltage of microscope was set to 20 kV. To prepare the samples for SEM analysis, nanofibers were directly electrospun on an aluminum foil. Image analysis software (ImageJ, National Institutes of Health, USA) was used to determine the nanofiber diameter distribution and morphology [22].
Transmission electron microscopy
Transmission electron microscopy (TEM) analysis was performed to prove and observe the size, shape and morphology of the core–shell electrospun nanofibers. To prepare samples for TEM analysis, formvar carbon-coated copper grids with 300 mesh was placed on the aluminum foil and then the nanofibers were directly electrospun on it. Nanofibers for the TEM image were analyzed by JEOL-1220 microscopy on an accelerating voltage of 80 kV.
Water contact angle
Water contact angle was performed to find out the hydrophilicity of the produced nanofiber membranes with the sessile drop method. Water contact angle was measured using an Attension range of contact angle meter and recorded by 10 µL water (for chromatography) volume that dropped onto the surface of the flat nanofiber membranes. Images were taken 10 s after the droplet had contact with the nanofiber mats.
Fourier Transform infrared spectroscopy
Fourier transform infrared (FTIR) spectroscopy of nanofibers was carried out using Perkin Elmar (Spectrum 100) to confirm the incorporation of OM oil into the nanofiber membranes. An Attenuated total reflection infrared spectroscopy (ATR-IR) spectrum was applied in the range of 4000–650 cm−1. To determine the spectral analysis, the samples were placed on a diamond crystal and FTIR spectrum was recorded.
Antimicrobial activity evaluations
Staphylococcus aureus (gram positive) and Pseudomonas aeruginosa (gram negative) microorganisms were used to determine the antibacterial activity of the produced nanofiber-based samples. The disk diffusion method was applied to the screening of antibacterial activity using 0.5 McFarland standard (1.5 × 108 CFU/mL) density suspension. The suspension was dispersed on a solid medium (Mueller–Hinton agar) and passed three times over the same area to make sure it was fully covered. Samples were placed on the petri plates and were incubated for 24 h at 37 °C temperature to measure the diameters of the inhibition zones. Circular pieces of nanofiber membranes were cut approximately with 6 mm in diameter for the antibacterial test.
In vitro drug release
For the drug release study, 5 mg of the nanofibers were immersed into a clean vial containing 5 ml of phosphate buffer solution (PBS, pH 7.4) with virtually no headspace. The samples were incubated at 100 rpm by a shaker at 37 °C temperature. At specified times, 1 mL of solutions were taken and transferred to a cuvette for analysis. After sampling, 1 mL fresh solution was added to it to maintain sink conditions [23]. The amount of released OM oil from the nanofibers containing OM was measured at 338 nm by employing UV–Vis spectroscopy (Thermo Electron Corporation) in the PBS solutions. The cumulative percentage release of OM from the nanofibers was calculated using the following formula [24]:
where C n determines the concentration of OM in the sample, V 0 introduces the volume of the release media, V r is the volume of the withdrawn sample, and W OM is the total amount of OM loaded into the nanofibers.
Results and discussion
Scanning electron microscope
The SEM images demonstrated that all the different target structures of the nanofiber mats have been successfully produced by the electrospinning technique. Moreover, no phase separation and delamination were observed in the structure of nanofibers. As displayed in Fig. 2a, virgin CH nanofiber membranes were formed into smooth, fine, continuous, consistent, and cylindrical shapes with an average diameter of 197 ± 10 nm. Adding OM oil to the polymer solution has led to obtain thicker nanofibers with an average diameter of 214 ± 11 nm (Fig. 2b, f) and broad size distribution range of approximately 100–400 nm. In comparison with pure CH, slightly rough and uneven surfaces were observed on the surface of B–OM nanofiber membranes. It verified the incorporation of OM into the CH polymer. In addition, polarity of chitosan and non-polarity of OM could lead to roughening of the surface, and the volume expands resulting in the formation of thicker nanofibers due to their inherent nature of tending to escape from each other. At the S–OM nanofiber membranes (Fig. 2c, g), OM loaded to the shell part of nanofiber membranes formed more patchy, rough surfaced and irregularly shaped nanofibers than B–OM nanofibers. The average fiber diameter for the electrospun S–OM nanofiber membranes was about 284 ± 12 nm with the size distribution range of 150–500 nm. Figure 2d shows a SEM image of the C–OM nanofibers wherein the nanofiber surface was relatively smooth and had a cylindrical-like shape like the pure CH nanofibers. The average diameter was almost similar with S–OM nanofibers of 291 ± 10 nm and the size distribution from 100 to 600 nm.

a–d SEM images of CH, B–OM, S–OM, and C–OM nanofibers membranes, and e–h demonstrate the nanofiber diameter distribution with the average diameter (n > 30), respectively
The SEM results suggested that the coaxial electrospinning process caused to increase the average diameter and fiber size distribution for the nanofibers when we compared with the single nozzle electrospinning process. Moreover, the results proved that when OM oil was incorporated into the shell layer as an active drug, the fibers became morphologically more rough, uneven and patchy-like structures. Whereas, while OM oil was embedded into the core layers, the fibers showed mainly smooth and regular surfaces.
Transmission electron microscopy
Figure 3 displays TEM images of produced C–OM and S–OM core–shell nanofiber structures under the optimized conditions. As realized, the core and shell structures of the nanofibers can be obviously seen. In the TEM images, we can observe a sharp boundary between the core and shell layers. The overall diameter of C–OM nanofiber membranes is 291 ± 10 with a core of 71 ± 7 nm (Fig. 3a). C–OM nanofibers were formed by uniform core layers in which the core layer of B–OM appeared darker than the shell layer of virgin CH polymer. On the other hand, the diameter of the S–OM nanofiber was about 284 ± 12 nm with the core part of 91 ± 7 nm (Fig. 3c). Interestingly, S–OM nanofiber membranes were composed of compact aligned dots which were encapsulated in the core layers. In this structure, capsules regions of virgin CH were lighter than the shell layer of B–OM. This feature affects the surface of the nanofibers as seen in the previous SEM images. The TEM observation confirmed that the OM oil has been incorporated into the electrospun nanofibers as the core and shell layers separately. To emphasize different structures of C–OM and S–OM, the schematic drawings are illustrated in Fig. 3b, d, respectively.

TEM images and schematic illustration of the core–shell structures of C–OM (a, b), and S–OM nanofiber membranes (c, d)
Water contact angle
As chitosan is known as hydrophilic in nature, the structure of the core–shell nanofibers after incorporation of OM can perform targeted and controlled delivery through postponing of penetrating the medium to the core layer [25]. The degree of hydrophilicity was determined by measuring the water contact angle of CH, B–OM, S–OM, C–OM nanofiber membranes as shown in Fig. 4. CH biopolymer indicates hydrophilic property due to the four hydroxyl group, a minor proportion of amide groups, with an amine group [26]. The average contact angle of the CH, B–OM, S–OM, C–OM nanofiber membranes are 40.68° ± 0.59°, 30.51° ± 0.057°, 32.92° ± 0.47°, and 37.17° ± 0.62°, respectively, after 10 s of contact the droplet had with the nanofiber membranes. Unexpectedly, the B–OM and S–OM nanofiber membranes were more hydrophilic due to the presence of OM oil in the blended form and in the shell layer. As previously stated, these nanofibers also possessed high antibacterial activity than the others. On the contrary, CH and C–OM nanofiber membranes indicated relatively less hydrophilic properties because of the existence of totally pure CH and OM in the only core layer. As a result, interestingly, electrospinning of the OM oil (non-polar) with chitosan enhanced the hydrophilic property of certain composite nanofibers. Indeed, at the beginning of the experiment, we expected to increase hydrophobicity with the addition of OM oil to the nanofibers. However, although the test was repeated three times, the hydrophilicity was increased by the order of B–OM, S–OM, C–OM, and CH. A different result was obtained than what we expected. During the experiment, we observed that CH started to swell and induced relatively fast diffusion of the entrapped OM oil from the nanofibers, where they were in contact with the water, to the outside, as we realized the same trend during the drug release behavior. After the diffusing, water can easily move into the nanofibers and tend to get it wet by spreading throughout the surface. The observation of oil in water emulsion on the surface of the nanofiber mat during the experiment is one of the verification to increase the hydrophilicity degree. Furthermore, in a study, the addition of essential oils to chitosan/poly(ethylene oxide) solutions resulted in decrease of the contact angle, and increase of hydrophilicity of the solution, because the oils lower the surface tension [27]. It can be another reason to obtain higher hydrophilicity for our nanofibers with the addition of OM oil.

Water contact angle measurements of the nanofiber structures
Fourier Transform infrared spectroscopy
In general, according to the literature, the antimicrobial activities of the compounds can be followed as phenols > aldehydes > ketones, and so on [28]. Phenol is an aromatic hydrocarbon group which has hydroxyl group bound on top as carbolic acid (C6H5OH) which is known to be one of the simplest phenolic compounds. In our study, it is expected that OM has two major phenolic components which are called carvacrol and thymol [29, 30]. The most intense bands for the components were observed in the IR spectrum at 804 cm−1 which is related to thymol and 811 cm−1 refers to carvacrol. In our spectra, the carvacrol band appeared at 813 cm−1 instead of 811 cm−1. These two bands are described as out-of-plane CH wagging vibrations [31]. Carvacrol and thymol mode of vibrations are stretching and bending, hence they represent an isomeric compound. The strong peak at 2962 cm−1 can be ascribed to CH stretching which are methyl and isopropyl groups on the phenolic ring [32]. In addition, carvacrol and thymol components have four specific peaks between 1622 and 1458 cm−1 which is related to the phenolic ring [32]. Other major bounds for carvacrol are 996, 1120, and 1176 cm−1 and for thymol are 946, 1088, and 1289 cm−1 [33].
The comparison of the FTIR results confirmed that our methods provides the opportunity for direct encapsulation of OM oil into the electrospun nanofibers of B–OM, S–OM, and C–OM at 996, 1461, and 2962 cm−1 peaks with a little shift as marked in Fig. 5.

FTIR spectra of CH, OM, B–OM, S–OM, and C–OM nanofibers
Antimicrobial activity evaluations
By applying the disk diffusion method, the activity intensity of nanofiber membranes can be visually checked by measuring the local inhibition zone [34] as Fig. 6 shows for S. aureus and P. aeruginosa. Based on the antibacterial results, we can conclude that OM oil increased the activity intensity of nanofiber membranes. Indeed, although the functionalized nanofiber membranes inhibited the growth of bacteria to some degree, the inhibitory zones were less than the control of amoxicillin activity.

Zone of inhibition of nanofiber membranes against S. aureus (a), and P. aeruginosa (b)
On the other hand, as seen in Fig. 6a, the inhibition zone for S. aureus bacteria can be visualized using B–OM and S–OM nanofiber membranes. The diameters of inhibition zones against S. aureus bacteria for B–OM and S–OM were 6 ± 1 and 9 ± 2 mm, respectively. However, CH and C–OM nanofiber membranes did not show any inhibition zone for S. aureus bacteria. CH has been known to possess antibacterial properties [35]. But, our study showed that virgin CH nanofiber membrane did not have any antibacterial activity against S. aureus bacteria. Recently, Akroun et al. [36] reported that chitosan-based nanofibers can inhibit the growth of S. aureus bacteria. This is in variance with our observation. The lack of antibacterial activity against S. aureus bacteria may be reasoned out by the following factors: polymer concentration, solvent type, molecular weight and the thickness of the nanofiber membranes [37]. In addition, some other studies demonstrate that the biocide effect of CH can be inhibited when CH is not able to diffuse through the adjacent agar [38, 39]. For our case, there might be a very limited diffusion of CH which was not enough for the antimicrobial activity and when OM oil was added to chitosan, it demonstrated a zone of inhibition due to the migration of OM oil or OM oil with a limited chitosan into the agar. Virgin CH polymer probably played as a barrier layer for OM diffusion to the outer surface of nanofibers to kill the bacteria when CH was incorporated to the shell layer during the testing time.
For P. aeruginosa bacteria, inhibition zone of CH, B–OM, S–OM, C–OM nanofiber membranes demonstrated 6 ± 0.0, 7 ± 0.0, 10.5 ± 1.5, 9 ± 0.0 mm, respectively. B–OM, S–OM, C–OM nanofiber membranes inhibited the growth of P. aeruginosa bacteria inside and around the nanofiber disks. Differences of the cell structures of P. aeruginosa and S. aureus might cause different effects of a very limited diffusion of CH through the agar. Therefore, it showed growth inhibition just inside the nanofiber disks without any inhibition zone around the disks for P. aeruginosa. In addition, there is no any significant difference between S–OM and B–OM for the antibacterial activity against S. aureus bacteria, but the difference is a slightly higher for P. aeruginosa probably due to the limited effect of chitosan. Figure 6b displays that the inhibitory effect increased after adding OM to the nanofiber membranes. As expected, S–OM nanofiber indicated the best antibacterial activity when compared with other functional nanofibers.
The antibacterial test result demonstrated that S–OM nanofiber membranes had more antibacterial activity than C–OM after 24 h against both the bacteria types. For the S–OM nanofiber membranes, probably the active OM in the shell layer was easily released into the environment [40]. In other words, the nanofibers surface and the shell part is more susceptible to exposure to the environmental conditions than the core part. The core part cannot be quickly dissolved or show release behavior in a short time, but it may display the release behavior over a long period of time [41, 42]. Thus, the nanofibers in which OM loaded to the core part did not show the same performance as S–OM for both the types of the bacteria during 24 h.
In vitro drug release
In general, chitosan possesses good water adsorption property. When the nanofibers are placed in the release medium, it is expected that the polymeric matrix started to swell and induce fast diffusion of the entrapped drug molecules from the nanofibers into the environment. In vitro drug release profiles of chitosan nanofibers using the single and coaxial electrospinning were investigated. The cumulative release behavior of the OM oil from nanofiber membranes is depicted in Fig. 7. The release profile can be studied in two stages: (1) an initial burst release, (2) constant slow release. For this research, the burst release was about 65, 47, and 28% for B–OM, S–OM, and C–OM nanofiber membranes, respectively, during the first 12 h. It is important for the medical treatment to prevent bacterial infection which is generally formed immediately after the incidents such as surgery and burn injury [43]. The release mechanism of OM oil from nanofibers possibly relates to relaxation of the polymers and diffusion behavior of the drug [44]. In this study, at the first 12 h, the highest initial burst release was observed especially for B–OM fibers about 65%, then followed by S–OM and C–OM nanofibers. Particularly, the core–shell nanofibers suppressed the initial burst release and provided a more sustained drug release profile in which the burst release at early times was reduced about 132% when B–OM and C–OM were compared.

Drug release profiles of the B–OM, S–OM, and C–OM nanofiber membranes
The results demonstrate that B–OM and S–OM nanofiber membranes have the similar drug release behavior during the first 7 h, and then started to differentiate between each other. It is known that S–OM did not have any OM in the core part, while OM dispersed to the whole structure of B–OM, therefore during the drug release, most probably some amount of OM diffused into the core part of S–OM instead of the outside environment. For this reason, it should be emphasized that only 85 and 71% of OM were released from B–OM and S–OM nanofibers and the rest stayed embedded even after the 4- and 6-day release study, respectively. These two groups of nanofibers can be used for the acute wound healing during hemostasis and inflammation phases.
Compared with those of B–OM and S–OM nanofiber mats, it is clearly indicated that C–OM had quite different and the slow drug release profile was the most probably due to the barrier effect of CH shell, which postpones the diffusion of OM out of the nanofibers [45]. As shown in Fig. 7, it can be observed that C–OM fibers have undergone the burst release of only 23% of its OM content within 8 h. And then the release profile becomes relatively slow and sustained for about 7 days. In addition, C–OM nanofibers demonstrated only 53% of the drug release after the 7-day release profile which renders the C–OM nanofibers efficient for sustained drug release. It can be used in such treatments which need delayed release profiles in the case of several diseases.
Conclusion
In this study, OM oil-loaded chitosan nanofibers were successfully fabricated by single and coaxial electrospinning for the core–shell structures as well as effectively evaluated for its efficiency to deliver the entrapped compounds. The C–OM nanofiber membranes showed uniform structure, while S–OM nanofibers indicated encapsulated unique bead-like structure in the core layers. As expected, the OM oil improved the antibacterial activity. Among all the nanofiber mats, S–OM exhibited the highest antibacterial activity comparing with the other membranes due to encapsulation of the active compounds into the shell layer. In the case of an in vitro drug release test, expectedly, the slowest drug release was observed for C–OM nanofibers during the entire period of time. This can be attributed to the delay of the diffusion of OM from the core part to out of the nanofibers.
As a result, production of different eco-friendly nanofeatured constructs of core–shell nanofibers can provide numerous implications and engineered to attain the required drug release profiles and antibacterial activities. They can be demanded by various medical applications from antibacterial to wound healing where burst or sustained drug release matters.
References
Huang Z-M, Zhang Y-Z, Kotaki M, Ramakrishna S (2003) A review on polymer nanofibers by electrospinning and their applications in nanocomposites. Compos Sci Technol 63(15):2223–2253
Sun Z, Zussman E, Yarin AL, Wendorff JH, Greiner A (2003) Compound core–shell polymer nanofibers by co-electrospinning. Adv Mater 15(22):1929–1932
Calisir MD, Erol M, Kilic A, Avci H (2016) Photophysical properties of phosphorescent elastomeric composite nanofibers. Dyes Pigm 125:95–99
Fang J, Niu H, Lin T, Wang X (2008) Applications of electrospun nanofibers. Chin Sci Bull 53(15):2265
Bhardwaj N, Kundu SC (2010) Electrospinning: a fascinating fiber fabrication technique. Biotechnol Adv 28(3):325–347
Pakravan M, Heuzey M-C, Ajji A (2012) Core–shell structured PEO-chitosan nanofibers by coaxial electrospinning. Biomacromol 13(2):412–421
Cheng L, Ma S, Wang T, Li X, Luo J, Li W, Mao Y, Gz D (2014) Synthesis and characterization of SnO2 hollow nanofibers by electrospinning for ethanol sensing properties. Mater Lett 131:23–26
Li Y, Lim CT, Kotaki M (2015) Study on structural and mechanical properties of porous PLA nanofibers electrospun by channel-based electrospinning system. Polymer 56:572–580
Hu X, Liu S, Zhou G, Huang Y, Xie Z, Jing X (2014) Electrospinning of polymeric nanofibers for drug delivery applications. J Control Release 185:12–21
Yarin A (2011) Coaxial electrospinning and emulsion electrospinning of core–shell fibers. Polym Adv Technol 22(3):310–317
Nguyen TTT, Chung OH, Park JS (2011) Coaxial electrospun poly (lactic acid)/chitosan (core/shell) composite nanofibers and their antibacterial activity. Carbohydr Polym 86(4):1799–1806
Sp Z, Sinha-Ray S, Sinha-Ray S, Kristl J, Yarin AL (2015) Long-term sustained ciprofloxacin release from pmma and hydrophilic polymer blended nanofibers. Mol Pharm 13(1):295–305
Siegel RA, Rathbone MJ (2012) Overview of controlled release mechanisms. In: Fundamentals and applications of controlled release drug delivery, Springer, New York, p 19–43
Boateng JS, Matthews KH, Stevens HN, Eccleston GM (2008) Wound healing dressings and drug delivery systems: a review. J Pharm Sci 97(8):2892–2923
Zahedi P, Rezaeian I, Ranaei-Siadat SO, Jafari SH, Supaphol P (2010) A review on wound dressings with an emphasis on electrospun nanofibrous polymeric bandages. Polym Adv Technol 21(2):77–95
Avci H, Guzel FD, Erol S, Akpek A (2017) Recent advances in organ-on-a-chip technologies and future challenges: a review. Turk J Chem. http://doi.org/10.3906/kim-1611-35
Avci H, Monticello R, Kotek R (2013) Preparation of antibacterial PVA and PEO nanofibers containing Lawsonia Inermis (henna) leaf extracts. J Biomater Sci Polym Ed 24(16):1815–1830
Rodriguez-Garcia I, Silva-Espinoza B, Ortega-Ramirez L, Leyva J, Siddiqui M, Cruz-Valenzuela M, Gonzalez-Aguilar G, Ayala-Zavala J (2016) Oregano essential oil as an antimicrobial and antioxidant additive in food products. Crit Rev Food Sci Nutr 56(10):1717–1727
Aslim B, Yucel N (2008) In vitro antimicrobial activity of essential oil from endemic origanum minutiflorum on ciprofloxacin-resistant Campylobacter spp. Food Chem 107(2):602–606
Ifuku S (2014) Chitin and chitosan nanofibers: preparation and chemical modifications. Molecules 19(11):18367–18380
Jayakumar R, Prabaharan M, Kumar PS, Nair S, Tamura H (2011) Biomaterials based on chitin and chitosan in wound dressing applications. Biotechnol Adv 29(3):322–337
Nurbas M, Ghorbanpoor H, Avci H (2017) An eco-friendly approach to synthesis and characterization of magnetite (Fe3O4) nanoparticles using Platanus orientalis L. leaf extrac. Dig J Nanomater Biostruct 12(4):993–1000
D’Souza S (2014) A review of in vitro drug release test methods for nano-sized dosage forms. Adv Pharm 2014:1–12
Che H, Huo M, Peng L, Ye Q, Guo J, Wang K, Wei Y, Yuan J (2015) CO2-switchable drug release from magneto-polymeric nanohybrids. Polym Chem 6(12):2319–2326
Karthikeyan K, Guhathakarta S, Rajaram R, Korrapati PS (2012) Electrospun zein/eudragit nanofibers based dual drug delivery system for the simultaneous delivery of aceclofenac and pantoprazole. Int J Pharm 438(1):117–122
Zhang H, Wu C, Zhang Y, White CJB, Xue Y, Nie H, Zhu L (2010) Elaboration, characterization and study of a novel affinity membrane made from electrospun hybrid chitosan/nylon-6 nanofibers for papain purification. J Mater Sci 45(9):2296–2304
Rieger KA, Birch NP, Schiffman JD (2016) Electrospinning chitosan/poly (ethylene oxide) solutions with essential oils: correlating solution rheology to nanofiber formation. Carbohydr Polym 139:131–138
Ballester-Costa C, Sendra E, Fernández-López J, Pérez-Álvarez JA, Viuda-Martos M (2013) Chemical composition and in vitro antibacterial properties of essential oils of four Thymus species from organic growth. Ind Crops Prod 50:304–311
Arumugam G, Swamy MK, Sinniah UR (2016) Plectranthus amboinicus (Lour.) Spreng: botanical, phytochemical, pharmacological and nutritional significance. Molecules 21(4):369
Pesavento G, Calonico C, Bilia A, Barnabei M, Calesini F, Addona R, Mencarelli L, Carmagnini L, Di Martino M, Nostro AL (2015) Antibacterial activity of Oregano, Rosmarinus and Thymus essential oils against Staphylococcus aureus and Listeria monocytogenes in beef meatballs. Food Control 54:188–199
Schulz H, Özkan G, Baranska M, Krüger H, Özcan M (2005) Characterisation of essential oil plants from Turkey by IR and Raman spectroscopy. Vib Spectrosc 39(2):249–256
Wu Y, Luo Y, Wang Q (2012) Antioxidant and antimicrobial properties of essential oils encapsulated in zein nanoparticles prepared by liquid–liquid dispersion method. LWT Food Sci Technol 48(2):283–290
Rodríguez-Solana R, Daferera DJ, Mitsi C, Trigas P, Polissiou M, Tarantilis PA (2014) Comparative chemotype determination of Lamiaceae plants by means of GC–MS, FT-IR, and dispersive-Raman spectroscopic techniques and GC-FID quantification. Ind Crops Prod 62:22–33
Ryan MP, Rea MC, Hill C, Ross RP (1996) An application in cheddar cheese manufacture for a strain of Lactococcus lactis producing a novel broad-spectrum bacteriocin, lacticin 3147. Appl Environ Microbiol 62(2):612–619
Goy RC, Britto D, Assis OB (2009) A review of the antimicrobial activity of chitosan. Polímeros 19(3):241–247
Arkoun M, Daigle F, Heuzey M-C, Ajji A (2017) Mechanism of action of electrospun chitosan- based nanofibers against meat spoilage and pathogenic bacteria. Molecules 22(4):585
Wang C, Duan L, Qin J, Wu Z, Guo S (2016) Studies on antibacterial activities against S. aureus of chitosan metal chelates prepared in magnetic field. J Appl Biomater Funct Mater 14(1):80–82
Rodríguez-Núñez JR, López-Cervantes J, Sánchez-Machado DI, Ramírez-Wong B, Torres-Chavez P, Cortez-Rocha MO (2012) Antimicrobial activity of chitosan-based films against Salmonella typhimurium and Staphylococcus aureus. Int J Food Sci Technol 47(10):2127–2133
Escárcega-Galaz AA, López-Cervantes J, Sánchez-Machado DI, Brito-Zurita OR, Campas-Baypoli ON (2017) Antimicrobial activity of chitosan membranes against Staphylococcus aureus of clinical origin. In: Enany S (ed) The rise of virulence and antibiotic resistance in Staphylococcus aureus. InTech, London, pp 109–124
Yan S, Xiaoqiang L, Lianjiang T, Chen H, Xiumei M (2009) Poly (l-lactide-co-ɛ-caprolactone) electrospun nanofibers for encapsulating and sustained releasing proteins. Polymer 50(17):4212–4219
Mickova A, Buzgo M, Benada O, Rampichova M, Fisar Z, Filova E, Tesarova M, Lukas D, Amler E (2012) Core/shell nanofibers with embedded liposomes as a drug delivery system. Biomacromol 13(4):952–962
Wang C, Yan K-W, Lin Y-D, Hsieh PC (2010) Biodegradable core/shell fibers by coaxial electrospinning: processing, fiber characterization, and its application in sustained drug release. Macromolecules 43(15):6389–6397
Ramakrishna S (2005) An introduction to electrospinning and nanofibers. World Scientific, Singapore
Wang X, Yue T, Lee T-C (2015) Development of pleurocidin-poly (vinyl alcohol) electrospun antimicrobial nanofibers to retain antimicrobial activity in food system application. Food Control 54:150–157
Yu H, Jia Y, Yao C, Lu Y (2014) PCL/PEG core/sheath fibers with controlled drug release rate fabricated on the basis of a novel combined technique. Int J Pharm 469(1):17–22
Acknowledgements
The authors would like to thank Scientific Research Projects Funds (BAP 2014–614) of Eskisehir Osmangazi University for the support of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Avci, H., Ghorbanpoor, H. & Nurbas, M. Preparation of origanum minutiflorum oil-loaded core–shell structured chitosan nanofibers with tunable properties. Polym. Bull. 75, 4129–4144 (2018). https://doi.org/10.1007/s00289-017-2257-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-2257-y