
Abstract
Methacrylamide-acrylic acid-N-isopropylacrylamide terpolymeric hydrogels have been prepared via free radical polymerization using diammonium-peroxo-disulphate initiator system and ethylene glycol-di-methacrylate as a cross-linker. The hydrogels were characterized by Fourier transform infrared spectroscopy, field emission scanning electron microscopy, thermogravimetric analysis, and differential scanning calorimetry. Molar mass was determined by viscometric method. Rheological profiling of the hydrogels was carried out by rheological measurements at various temperatures and methacrylamide contents in hydrogels. Thermogravimetric data were analyzed to assess the kinetic triplet by iso-conversional method. The values of apparent activation energy (E a) figured out from TG data were measured for each step by Kissinger–Akahira–Sunose method. On the findings of results, the decomposition reaction of terpolymeric hydrogels has been analyzed as a slow reaction, which is evident from the low-frequency factor values (A), i.e., 3 × 106, 2 × 1010, and 4 × 1015 s−1, respectively. The master plot method was used to predict the mechanism for degradation of MANIPAM-7. Flow curve and frequency sweep tests at different temperatures showed temperature dependency with shear rate indicating the pseudo-plastic nature of the synthesized samples. The rheological parameters were analyzed using various models. Out of these methods, the Ostwald, modified Bingham, and Herschel–Bulkley models were established to provide better fit to experimental data. Dynamic moduli measured as a function of angular frequency showed viscoelastic nature of all the samples. Frequency sweep studies indicated that storage (G′) and loss modulus (G″) showed an increase with increasing concentration of methacrylamide. Composition dependency of crossover frequency directed an increase in relaxation time with methacrylamide contents in the hydrogel.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The role of pharmaceutical and industrial revolution in human society is intensifying every day. Polymer materials are enormously versatile and becoming progressively more admired in all areas of modern life. There are many grades of plastics available to today’s designer with novel and innovative formulations developing daily [1]. Because of diversity and wide range of applications, it accentuates researchers to design and synthesize such a peculiar, innovative, and high-performance material to meet the daily demands of human life. Polymer science is an interdisciplinary area consisting of chemical, physical, engineering, processing, and theoretical aspects. It also has enormous impact on contemporary materials science. Numerous modern functional materials and devices have polymers as primary parts. Not surprisingly, approximately 30% of all scientists in the chemical industry work in the field of polymers. The polymer science is of ever-increasing significance to everyone’s daily life [2]. Different types of polymers exist in nature. Among those are starches, silk, asbestos, sand, and cotton, as well as the incredibly complex polymers known as RNA (ribonucleic acid) and DNA (deoxyribonucleic acid), which clutch genetic codes. Artificial polymers include plastics such as styrofoam, polyethylene, Saran wrap, Dacron (polyester), and rayon, and fibers such as nylon and other materials such as PVC pipe, Formica, and Teflon [3, 4]. Polymers can be used as stabilizers, solubilizers, and mechanical supports for sustained release of drugs. But over a long run, the functionalities and performances of polymers have commutated. Now a day, polymers are used as insulators and resistant to heat and chemicals.
In addition to these properties, polymers also have ability to imbibe large amount of water. This distinctive kind of polymers is referred to as hydrogels. They are cross-linked three-dimensional hydrophilic polymer networks that swell up, but do not dissolve when brought into contact with water. The hydrogel is a class of polymeric materials with the capability to hold a sizeable amount of water, presenting a soft, rubbery-like consistency, and low interfacial tension parameters. Their properties mainly depend on the chemical composition of the polymeric chains, the extent of cross-linking, and the interaction between the network and surrounding liquids.
Hydrogels undergo volume changes reversibly in response to external and foreign stimulus such as temperature and pH, hence are called as SMART hydrogels [5]. During the last two decades, the natural hydrogels are deliberately taken over by synthetic hydrogels which have long working life, high gel strength, and high water absorption capacity. Synthetic polymers have well-defined structures that can be altered to yield tailorable degradability and functionality in the polymer [6]. So due to some unique and particular properties such as elasticity, high water content, and capacity to control these properties, they are useful for various applications including wound dressing, contact lenses, tissue engineering, and drug delivery systems.
In drug delivery systems, hydrogels can control the release of drug in response to the environmental stimuli by altering its structure and also have the ability to shield and protect the drug from unfriendly environments. These gels have a high degree of flexibility very similar to natural tissues due to their large water contents. The pH-sensitive hydrogels can be synthesized usually by adding some pendant basic or acidic functional groups in the polymer backbone. They can either release or accept protons in response to suitable pH. These gels have network permeability which changes with electrostatic repulsion. Contrariwise at low pH, the hydrogels with basic pendant groups such as amines show electrostatic repulsion.
Copolymeric hydrogels of N-isopropylacrylamide-co-acrylic acid have already been reported [7]. These hydrogels are pH sensitive with anionic acrylic acid and non-ionic acrylamide. The carboxylic and amide moieties in the polymer backbone impart appropriate polarity and hydrogen bonding for hydration. For the preparation of pH-sensitive hydrogels, acrylic acid, methacrylamide, and N-isopropylacrylamide monomers were selected. The swelling of gels depends on acrylic acid, which is expedited by the presence of carboxylic groups in the polymer network which are powerfully associated with water molecules. These functional groups are easily ionisable and highly sensitive to the effects of ionic strength and pH. Acrylic acid can swell in water and show volume phase transition with changing pH at around pH 7.4 [8]. In addition, in acidic medium, its swelling activity will be low as compared to basic medium. Hence, on loading the drug in the hydrogel, the drug release will be in alkaline pH, but the drug will be sustained in acidic media, i.e., stomach. While acrylamide is a hydrophilic monomer, its homo-polymer does not display any pH-induced volume phase transition in water.
The homo-polymer of N-isopropylacrylamide is a thermo-responsive polymer showing a volume phase transition at 306 K (LCST) [6]. Below 306 K, the hydrogel is hydrophilic, hydrated, and swollen while above this temperature, the hydrogel is dehydrated, shrank, collapsed, and shows hydrophobicity due to the cleavage of the hydrophilic/hydrophobic equilibrium in the linked structure. This unique property of NIPAM is due to its quick repetition in hydrophobicity and hydrophilicity around LCST.
To investigate the thermal stability of hydrogels, thermogravimetric analysis had been generally employed for kinetic degradation study, which is responsible for their applications in various fields [9]. The TG study at different temperatures gives various information about nature of the species formed during the degradation process. Various kinetic and thermodynamic parameters determined by this technique are apparent activation energy (E a), frequency factor (A), reaction mechanism g(α), entropy change (∆S*), Gibbs free energy change (ΔG*), and enthalpy change (∆H*).
So, it is the rheology that decides whether a polymer can be formed, processed, and shaped into a desired article in an efficient and economical approach, while maintaining the dimensional stability and high quality. Therefore, rheological assessments are very imperative for characterization of a hydrogel because the mobility required for drug diffusion can be indirectly investigated using these measurements. Extensive study of these measurements may lead to probable employment of different rheological models and parameters for the optimization of drug delivery from dermatological formulations. To obtain information about the microstructure, elastic, and viscous behavior of these gels, oscillatory rheometric and flow viscometric measurements were conducted. Experiments were performed to analyze the deformation, stability, and development of the microgel network through a varied range of flow and deformation conditions [10].
The present study focuses on the synthesis of methacrylamide-acrylic acid-N-isopropylacrylamide terpolymeric hydrogel. The introduction of acrylamide and its derivatives with the following properties, i.e., non-toxic, long chain length, biologically inert, and quality to preserve their shapes improve the mechanical strength, pH sensitivity, physical, and chemical properties of these hydrogels.
The aim of our work is to investigate the viscoelastic properties, i.e., zero shear viscosity (η o ), storage (G′) and loss modulus (G″) of the gels, yield strengths (τ o ), and determination of kinetic triplet [apparent activation energy (E a), frequency factor (A) and mechanism of thermal degradation] of polymer materials. Thermodynamic parameters, i.e., Gibbs free energy change (∆G*), enthalpy change (∆H*), and entropy change (∆S*) were also evaluated.
Experimental
Chemicals
The chemicals used in the present investigations are as follows: ethanol (BDH, 99.8%), diammonium-peroxo-disulphate (Fischer scientific, 98%), acrylic acid (Fluka, 99%), methacrylamide (Sigma-aldrich, 98%), N-isopropylacrylamide (Sigma-aldrich, 97%), ethylene glycol-di-methacrylate (Aldrich 95%), and DMSO (Applichem, 98%).
Synthesis of methacrylamide-acrylic acid-N-isopropylacrylamide (MAA-AA-NIPAM) terpolymeric hydrogel
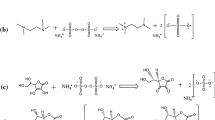
A series (MANIPAM-1 to MANIPAM-7) of titled polymeric hydrogels were prepared with variant contents of methacrylamide (0.00176, 0.00294, 0.035, 0.00528, 0.00587, 0.0088, and 0.0129 mol, respectively) while keeping other monomeric contents constant (0.015 mol acrylic acid, 0.0097 N-isopropylacrylamide mol, and 0.0053 mol EGDMA) for each sample. In a typical experiment, acrylic acid (0.015 mol), N-isopropylacrylamide, (0.0097 mol), and methacrylamide (0.00176 mol) were mixed in a beaker (25 mL) containing 5 ml of ethanol as a solvent. The reaction mixture was sonicated for 30 min to ensure the homogeneity of solution. To this reaction mixture EGDMA (0.0053 mol) and diammonium-peroxo-disulphate (1% w/v) were added as a cross-linker and as an initiator, respectively. Whole contents of the beaker were transferred into a screw capped tube and nitrogen gas was bubbled for 15 min through the reaction mixture to provide inert atmosphere. The tube was capped immediately and kept in a water bath preset at 308 K. After 6 h, the reaction mixture became viscous and its thickness increased with aging. Finally, the reaction mixture was further left for 24 h to ensure the completion of the polymerization reaction. The pale yellow final product was removed from the tube, washed with ethanol (2 × 5 mL), and deionized water (2 × 5 mL) to get rid of unreacted material and the solvent (Scheme 1). The samples were dried in an electric oven at 60 °C for 6 h. The synthesized sample was labeled as MANIPAM-1. Same procedure was adopted for the synthesis of other samples and preserved for further investigations.

Schematic representation of the synthesis of methacrylamide-acrylic acid-N-isopropylacrylamide terpolymeric hydrogel (MAA-AA-NIPAM)
Fourier transform infrared (FTIR) spectroscopy
In the present study, the FTIR spectrum of MANIPAM-7 hydrogel was recorded using NICOLET 6700 FTIR spectrophotometer, Thermoscientific USA.
Field emission scanning electron microscopy (FESEM)
The surface morphology of the synthesized samples MANIPAM-5 and MANIPAM-7 were visualized using “JEOL-JSM-6700F Field Emission Scanning Electron Microscope” (FESEM).
Molar mass determination
Molar mass of the polymeric hydrogel MANIPAM-7 was estimated utilizing viscometric data obtained from “Modified Ubbelohde Viscometer tube” with capillary section size (0.6–0.8 mm) by Sigma-Aldrich.
Thermogravimetric analysis (TGA)
Thermal degradation of the hydrogel MANIPAM-7 was carried out by thermal analyzer Universal 4.2E (TA instrument, USA) under nitrogen atmosphere at four different heating rates, i.e., 5, 10, 15, and 20 K/min. The data were obtained in a temperature range from ambient to 873 °C. The TG was used for the determination of various kinetic and thermodynamic parameters by iso-conversional method.
Differential scanning calorimetry (DSC)
DSC scans of two polymeric samples MANIPAM-5 and MANIPAM-7 were carried out using TA instrument coupled with SDT Q600 V20.9 Build 20 DSC under N2 atmosphere. Heating rate was about 10 K/min. The parameters such as glass transition temperature (T g), crystallization temperature (T c), and melting temperature (T m) were determined using this technique.
Rheological measurements
In the present study, the rheological measurements, i.e., flow curve tests and frequency sweep test were performed with a Physica MCR-301 (Anton Paar) Germany rheometer. The measurements were made at five different temperatures (293, 303, 313, 323, and 333 K). Before taking each measurement a pressure of 5 bar was maintained, a parallel plate (PP25/PP50) with a 1 mm diameter was attached to the rheometer. The required temperature for each measurement was attuned and a gap between plates was set at a distance of 0.125 mm. For flow curve test, the parameters such as shear stress and viscosity were recorded as a function of shear rate in the range 0.01–100 s−1. Frequency sweep test for all the samples was performed to evaluate complex viscosity (η *), storage, and loss modulus (G′ and G″) as a function of angular frequencies in the range 0.5–500 s−1.
Result and discussion
FTIR spectrum
Figure 1 represents the FTIR spectrum of MANIPAM-7 scanned in the range 400–4000 cm−1. The characteristic broad band due to (O–H) appeared at 3130 cm−1 with a shoulder due to (N–H) stretching at 3414 cm−1. Broadness of band indicates the existence of hydrogen bonding between the groups. Peak observed at 1715 cm−1 indicates the presence of carbonyl (C=O) stretching vibration of ɑ–ß unsaturated ester. The bands observed at 1650 cm−1 relates to the stretching vibration of the carbonyl (C=O) of amide group. The characteristic peak at 1404 and 1190 cm−1 are due to the presence of C–C (1300–1450) bending vibrations and C–N (1000–1260) stretching vibration, respectively. The peak recorded at 1047 cm−1 is assigned to the stretching vibration of (C–O–C) group which reveals the presence of EGDMA. The band below 900 cm−1 belongs to the different bending vibration of C–H bond. The IR data indicate the formation of the target polymer.

FTIR spectrum of MANIPAM-7
Field emission scanning electron microscopy (FESEM) analysis
Morphological examination of synthesized polymeric gels MANIPAM-5 and MANIPAM-7 is presented in Fig. 2. It is clear from Fig. 2a that the hydrogel has entangled three-dimensional molecular networks showing that the chains are intertwined and are not independent and individual. These chains are made up by the combination of particles with clear boundaries and are almost round in shape. The average particle size is in the range 0.3–0.6 μm.

SEM micrographs for polymeric sample MANIPAM-5 (a) and MANIPAM-7 (b)
SEM images showed that the chains, although entangled with one another, are not coiled up. It has been observed that the cross-linked chains with less methacrylamide concentration have empty spaces between the chains while with greater concentration of methacrylamide these chains show garlic-like structure (Fig. 2b), and the size of the particles is in the range 1–5 μm. It can be interpreted that with increasing concentration of methacrylamide the entanglement between the chains is increased.
Molar mass determination
To characterize polymeric gels, molar mass is an important parameter. Some methods cannot fractionate the sample and for all the molecules they provide only averages of their molecular weights. These average masses have numerous forms, viz., number average (\(\bar{M}\) n ), weight average (\(\bar{M}\) w ), viscosity average (\(\bar{M}\) v ), and z-average (\(\bar{M}\) z ). These averages are mathematically expressed as follows:
where M i is the molar mass having n i number of molecules.
It has often been found that weight average (\(\bar{M}\) w ) and viscosity average (\(\bar{M}\) v ) are close to each other. Therefore, the weight average (\(\bar{M}\) w ) can be determined using viscometry technique. In the present studies, the weight average molar mass of the synthesized polymer was determined from viscometric data using Mark–Kuhn–Houwink–Sakurada equation:
where (K) and (ɑ) are characteristic constants which depend on the solvent and temperature. For DMSO, (ɑ) is 0.610 and (K) is 0.001710 at 298 K. Intrinsic viscosity of the polymer was evaluated as intercept of Huggins plot (Fig. S1) (using viscometric data obtained from “Modified Ubbelohde Viscometer tube”). The molar mass calculated for the polymeric gel was of the order of 1.13 × 105 g/mol. This value indicates that the synthesized sample may be considered as high molecular weight polymer.
Thermogravimetric analysis
Thermal decomposition pattern
Terpolymeric hydrogels can be thermally degraded and their degradation patterns can be studied during heating by defining their mass loss curves. The representative TG and DTG curves of polymeric sample MANIPAM-7 at four different heating rates, i.e., 5, 10, 15 and 20 K/min are shown in Fig. 3a, b. The thermograms of polymeric gels showed three decomposition stages. In the first stage, the weight loss is about 12.32% in the temperature range 345.6–485.2 K due to small amount of initiator, ethanol, and water evaporation in polymer matrix. In the second stage, 50.008% weight loss over the range (485.2–679 K) may be related to the degradation of polymer backbone [11]. The third decomposition stage (679–828 K) with 36.667% weight loss showed the decomposition of residual polymeric chains and branches in the structure [12]. From TGA, it is revealed that the polymeric gels gain more thermal stability at higher heating rates. The second stage of degradation with 50% weight loss may be considered as a major degradation step.


a Representative plots of temperature versus weight loss of MANIPAM-7 at 5, 10, 15, and 20 K/min, b differential thermogravimetric (DTG) curves of MANIPAM-7 at 5, 10, 15, and 20 K/min
Thermal stability at various heating rates
The representative (α–T) curve for second (major) step of thermal degradation is shown in Fig. S2. It has been observed that the mass loss at higher heating rate occurs at higher temperature which advocates the high thermal stability of sample at higher heating rate. It can be explained in such a way that with the variation of heating rate, heat transfer rate also varies. Thus, the sample experiences a short exposure time for a typical temperature [13]. As an outcome of the rapid heating, the exterior surface of sample gets hotter than its core. As a result the reaction occurring in interior of the sample will take place at lower temperature. However, the product thus obtained has to cross high temperature region where they may undergo secondary reactions. Similar trends have also been reported by Jaber et al. [14].
Determination of kinetic triplet
Thermogravimetric data were analyzed for the determination of kinetic triplet. The progress of decomposition reaction was assessed by determining the value of α at a particular temperature:
where m i, m f, and m t refer to initial, final, and mass at any given temperature, respectively.
Apparent activation energy (E a)
The apparent activation energy (E a) for all the steps is calculated by Kissinger–Akahira–Sunose method (KAS).
where T p is the peak temperature (the temperature corresponding to maximum weight loss) and β is the heating rate. The values of E a obtained from slope of the line (Fig. S3(a)) for each degree of conversion (a) value are listed in Table 1.
From Table 1, it is clear that the E a for all the three steps is of the order
Third step values are higher in magnitude than the former two steps which indicate the relatively higher stability of the polymer as compared to its degradation in the former two stages.
Reaction mechanism
The reaction mechanism g(α) was determined by master plot method:
The theoretical master plots were determined by plotting g(α)/g(0.5) against (α). Here, g(α) represents various models (Table S1) which control the degradation process, while g(0.5) is the g(α) value for a reference point α = 0.5 [15]. Similarly, the experimental master plots were obtained by plotting p(x)/p(0.5) and (α). In current studies, p(x) has been achieved by using Senum–Yang’s fourth degree approximation:
where x = E a / RT and p(0.5) is the p(x) for a reference point when α = 0.5. So the plot of (α) versus g(α)/ g(0.5) represents the theoretical master plot, while the plot of p(x)/p(0.5) versus (ɑ) gives experimental master plots. For a given g(α), if the theoretical plot shows best matching with the experimental plot then it will be the required mechanism of the thermal degradation reaction. The results of this analysis (Table 1) showed that the first degradation step followed contracting sphere model (R 3, F 2/3 model for α = 0.1–0.9), which is a contracting sphere model. According to this, model nucleation takes place on the crystal surface and this phenomenon is very rapid. So, the degradation rate can be controlled near the center of the crystal by the progress of the resulting reaction occurring at the interface [16].
Second and third thermal degradation steps are well described by P 1/4 mechanism which follows Mampel power law model. The representative plot for second step of thermal degradation is shown in Fig. S3(b). This model is one of the models which include acceleratory rate equations. The rate determining mechanism for this degradation step is nucleation (the process in which new structures or new thermodynamic phases are formed via self-organization) [17].
Reconstruction of g(α) model
To confirm the reaction mechanism, g(α) model for all the steps was reconstructed using Eq. 7c. All the reconstructed plots confirmed the discussed reaction mechanisms. Representative plot of reconstructed model for second step is given in Fig. S3(d).
Evaluation of frequency factor (A)
The frequency factor (A) is determined by the slope of the plot of g(α) versus E*p(x)/βR.
where g(α) represents the different theoretical models presented in Table S1. The values of kinetic triplet, i.e., apparent activation energy (E a), frequency factor (A), and model of thermal degradation g(α) for all steps are presented in Table 1.
The representative plot of frequency factor (A) for first step is shown in Fig. S3c. The predictable values of (A) for three-step reaction are given in Table 1. It can be seen from the table that the values of (A) for first thermal degradation stage of reactions are low which indicates that reactants can rotate freely and the activated complex is restricted representing the formation of a tight complex [18]. This interpretation is in line with the argument given by Vlaev et al. [19], while comparing the kinetic results of non-isothermal degradation of calcium oxalate monohydrate. The second and third steps of thermal degradation have higher values of frequency factor as compared to the first step demonstrating the formation of loose complex [19]. Therefore, it is evident that activated complex is free to rotate parallel to the surface unlike the reactants.
Thermodynamic parameters
Thermodynamic parameters such ΔS*, ΔH*, and ΔG* were calculated for thermal degradation of polymeric sample using Eqs. (8–10) and their values are given in Table 2.
It can be seen from the table that the value of ΔG* gradually increases from first and third step of polymer degradation indicating that as the degradation reaction proceeds it becomes more and more non-spontaneous. The ΔH* specifies the energy difference between reactants and the activated complex. Small value of ΔH* for first step represents that the activated complex will be formed faster due to small potential energy barrier [20]. In the present study, ΔH* values for second and third steps are higher in magnitude than first step which indicate that the potential energy barrier shows an increase due to larger difference between reactants and the activated complex.
Georgieva et al. [20] reported that ΔS* reveals the closeness of the system in the direction of its own thermodynamic equilibrium. In the present study, the ΔS* values are small correlates to the physical or chemical changes in the reactants which takes the system near to its own thermodynamic equilibrium. It means in this case, reactants have low reactivity values which indirectly enhance the time required for the formation of an activated complex. Moreover, in the present study, the ΔS* values have negative sign which revealed that the structure of activated complex is well ordered relative to initial reactants, i.e., they had lost their translational degree of freedom. Such types of reactions can be termed as slow reactions [21].
Differential scanning calorimetry (DSC)
Different phase temperatures such as glass transition temperature (T g), crystallization temperature (T c), and melting temperature (T m) of the two polymeric gels MANIPAM-5 and MANIPAM-7 were determined using DSC analysis. DSC curves are shown in Fig. S4(a–b). These curves illustrate three distinct stages represented as an endothermic peak (glass transition temperature), an exothermic peak (crystallization temperature), and another endothermic peak representing the melting temperature, respectively. It was generally understood that below the glass transition temperature, the Brownian macromobility of the molecules is primarily controlled by intermolecular interactions, which show the existence of only restricted thermally induced movements of chain segments and negates any position change process in the molecule. Whereas the Brownian micromobility of the polymeric chains begins at this (T g), making the plastic soft, it did not affect the mechanical stability of polymeric chains [22]. A careful examination of (T g) temperatures of both samples showed that glass transition temperature (T g) of MANIPAM-7 at 462.89 K (189.89 °C) was higher with greater contents of methacrylamide as compared to MANIPAM-5 having T g at 458.79 K (185.79 °C). It can be explained in such a way that the extent of cross-linking in case of MANIPAM-7 was higher than that of MANIPAM-5, subsequently the segmental movement in the former case was more restricted. It was also observed that softening of the solid sample takes place at higher temperature in the case of MANIPAM-7 than that of MANIPAM-5.
However, crystallization temperature (T c) for both samples obtained in the present studies show no significant change. The discrepancy in melting temperature (T m) of these two samples was greater, but in reverse order. Low value of (T m) in the case of MANIPAM-7 suggested that as methacrylamide contents increased, the molecular ordering and the crystallinity of the polymer decreased. It can be attributed to an intermolecular interaction (hydrogen bonding) between the components in the polymer [23].
Rheological properties
Flow curve
The rheological properties of polymeric samples with different concentrations of methacrylamide were studied by oscillatory measurements. Shear stress (τ) and apparent viscosity (η) of synthesized samples MANIPAM-1 to MANIPAM-7 were plotted at five various temperatures as a function of shear rate. A representative flow curve of MANIPAM-1 at 293 K is shown in Fig. 4. It is clear from the figure that shear stress increases with increase of shear rate and same trend was observed for all the synthesized samples. This may be due to enhanced intermolecular interactions [24], whereas the viscosity shows initially a minute decrease with increase of shear rate as well as temperature. It is apparent from Fig. 4 that at low shear rate, a slight shear thickening behavior is observed followed by shear thinning at higher shear rates.

Representative plot of apparent viscosity and shear stress as a function of shear rate of MANIPAM-1 at 293 K
This trend of shear thickening followed by shear thinning can be interpreted in such a way that at low shear rates, the polymeric stretching chains may form aggregates which shows resistance in the flow and increases the viscosity of polymeric system. As the shear rate is further increased, these aggregates may collapse thus making the flow of the macromolecules easy, resulting in a decrease in apparent viscosity [24]. Furthermore, it was observed that when rotational speed is increased the alignment of molecules also increases in the direction of flow, resultantly the resistance is decreased in the flow and hence the decrease in viscosity is observed.
Temperature dependence of shear viscosity
For fluids having non-Newtonian behavior, the viscosity is considered at some specific shear rate to estimate the temperature dependency of viscosity. In the current study, 1.0 s−1 shear rate was applied to evaluate this dependency of viscosity on temperature. All the polymeric samples viscosities for a temperature range from 293 to 333 K are given in Table S2. The data show that the viscosity of all the samples decreases with increase in temperature indicating the non-Newtonian behavior. The decrease in viscosity was rapid at low shear rates (less than 100 s−1) while it was slow at high shear rates (more than 200 s−1). This decline in viscosity may be the consequence of reduction in entanglement between the macromolecular chains and viscosity of the solvent at higher temperature [25]. Furthermore, this fall in viscosity may also be attributed to the availability of greater free volume around the polymer molecules and provision of minimum thermal energy, due to higher temperature, and to overcome hindrance in the flow.
Composition dependence of zero shear viscosity (η o)
It is interesting to note here that the rheological properties of terpolymeric gels depend upon the methacrylamide concentration. Plot of viscosity versus shear rate gives straight lines whose intercepts gave the value of zero shear viscosity (η o). The (η o) values for all the samples studied at five temperatures are given in Table S3. From the table, it is clear that (η o) is found to decrease with increase in temperature, but increase with increase in concentration and this effect is more prominent in those polymeric samples which have high methacrylamide concentration. It may be ascribed that these samples have higher intermolecular associations rather than those having lesser amount of methacrylamide. Such behavior can be clarified due to the existence of entanglements in long polymeric chains within their structure, which remains uninterrupted at small shear. Al-Fariss and Al-Zahrani [26] have reported some observations similar to this.
Flow curve modeling
For the evaluation of rheological parameters, viscometric data were subjected to various mathematical models. Power law model (Eq. 11a–11b) was applied to conclude the value of consistency coefficient (K) and fluidity index (n), whereas yield stress (τ o) values for the synthesized samples were determined by modified Bingham (Eq. 12a–12b) and Herschel–Bulkley models (Eq. 13). Values of these parameters obtained are listed in Table 3.
Power law model
This depicts the fit of (Fig. S5a) Ostwald model (Eq. 11a) and Ostwald–de Waele model (Eq. 11b) to the rheological data of MANIPAM-1 at 323 K.
where (K) is consistency coefficient, (n) is the fluidity index, (γ) is the shear rate, (τ) is shear stress, and (η) symbolizes viscosity.
The values of consistency coefficient (K), fluidity index (n), and correlation coefficient (R 2) of all the samples for both models are given in Table 3. Based upon (R 2) values, it is concluded that Ostwald model provides better fit to the experimental data well. The values of (n) as evidenced from Table 3 are less than 1.0, advocating the pseudo-plastic nature of the synthesized polymeric samples [27]. It was further observed that the value of (n) changed to smaller value with increasing contents of methacrylamide. So, the polymer having higher contents of methacrylamide exhibits more non-Newtonian behavior. The probable reason for this trend may be the increase in intermolecular interactions due to higher contents of methacrylamide. However, the values of consistency coefficient (K) decreased with increase in temperature but increased with increasing polymer concentration (Table 3). The greater value of (K) at lower temperature reveals higher degree of entanglement between polymeric chains. As temperature increases, entanglement is broken down making the consistency coefficient smaller [28].
Yield stress determination
Minimum stress essential to make the material really flow, being associated with the internal structure of the material that necessarily be broken, is referred to as yield stress. Among various models used for yield stress determination, Casson model cannot be applied to the present data, because it assumes value of n = 0.5 [29]. So, due to clear physical meaning and the simplicity Bingham (Eq. 12a) and modified Bingham models (Eq. 12b) were applied to experimental data. Modified Bingham model accounts for the curvatures at low shear rates of the rheological curves to calculate the yield stress (τ o). The representative plot of Bingham and modified Bingham model for MANIPAM-7 at 303 K is demonstrated in Fig. S5b.
where τ o, τ, γ, and μ are yield stress, shear stress, shear rate, and plastic viscosity, respectively. At high shear rates the plots of τ–γ relationship are linear and they can be extrapolated to zero shear rate, where intercept gave the value of yield stress (τ o). The yield stress (τ o) values obtained for all the samples at all the studied temperatures are listed in Table 3. By comparing R 2 values of both models, it can be inferred that modified Bingham model fits better to the experimental data for all polymeric samples at all temperatures. The obtained (τ o) values in current studies (Table 3) are in the range 0.1–16.81 Pa. Similar observations have also been reported for some other hydrogels [30].
Herschel–Bulkley model
The Bingham model includes together the limiting viscosity (μp) and a yield stress (τ o) at definite shear rates, whereas the power law model does not consider them. Shear thinning and shear thickening behaviors can be observed in a non-linear flow relationship and the supposition that plastic viscosity value becomes constant is not valid. According to this model, fluid can be mathematically represented as follows:
where τ o , τ, K 1 , n, and γ represent the yield stress, shear stress, correction parameter, flow behavior index, and shear strain, respectively. τ o , n, and K 1 values were obtained by non-linear regression and are given in Table 3.
From the table, it is clear that the yield stress values exhibit temperature dependency. The yield stress and the flow behavior index values were decreased with increase in temperature. This drop in yield stress values can be explained with the help of Herschel–Bulkley model that the flow was homogenous for all shear rates, but it was not the case in this polymeric sample at low shear rates. It can be named as due to slip effects [31].
Frequency sweep studies
Complex viscosity
Polymers have different frequency-dependent properties. Complex viscosity (η*) is one of them which can be measured during involuntary harmonic oscillation. Oscillatory frequency sweep test was carried out to determine the viscoelastic behavior of the synthesized polymeric samples (MANIPAM-1 to MANIPAM-7). A representative composite plot of complex viscosity of the sample as a function of angular frequency at various temperatures is shown in Fig. 5.
It was observed that the complex viscosity exhibits decline with increasing angular frequency showing a shear thinning for all polymeric samples at all the investigated temperatures. Similarly, a smooth decrease in complex viscosity was examined as the concentration of methacrylamide increased in the polymeric gel. The decline in complex viscosity can be explained on the basis of entanglements in the polymeric chains which requires more time to relax at low deformation rate and this relaxation time can be decreased by increasing the sweeping frequency which finally leads to a decrease in complex viscosity.
Many structural changes can be seen at such higher frequencies by making and breaking of the molecular bonds than can be irreversible which leads to disentanglement of long polymeric chains and consequentially the complex viscosity is decreased. However, a certain frequency after these polymeric chains may recombine shows increase in complex viscosity. This kind of behavior is known as shear thickening. Similar observations have also been reported by Yurri et al. [32]. However, Wang predicted that when there are free chains in the existing network, they may coagulate and resultantly an increase in complex viscosity is observed showing the shear thickening behavior.
Dynamic moduli
The cross-linking kinetic information can be obtained through dynamic rheological routes. It is done by knowing the crossover point throughout the reaction and also by the polymer concentration effect on viscoelastic properties of the polymers. The characteristic mechanical properties of the gel can be deliberated in the form of elastic modulus (G′) and viscous modulus (G″), where (G′) and (G″) reflect the reversibly stored energy of the system and irreversible energy loss, respectively. In the present study, it has been observed that the storage modulus (G′) and loss modulus (G″) of all the polymeric samples increase with angular frequency at all the temperature ranges studied. A representative plot of angular frequency dependence of storage modulus (G′) and loss modulus (G″) of the MANIPAM-1 at 293 K is presented in Fig. 6.

Representative composite plot of complex viscosity as a function of angular frequency at different temperatures
In these curves, shear modulus represents the Maxwellian behavior showing the variations of G′ and G″. It is clear from Fig. 5 that at low angular frequency (ω) region, (G″) was greater than (G′) which indicates the liquid-like nature of these samples [33]. However, when frequency was further increased both moduli show an increase and finally cross each other at a particular frequency known a crossover frequency (G c). At this frequency, these curves show deviations from Maxwellian behavior representing the presence of faster relaxation times, and show superimposition in slow relaxation processes. The interpretation of this type of behavior can be described in terms of imperfections present in macromolecular microstructures and the polydispersity of these terpolymers, which represent the broadening of relaxation spectra. After this crossover frequency (G c), (G′) and (G″) show variation over the frequency where (G”) was decreased and (G′) was increased at higher angular frequency (ω) indicating the solid-like behavior (domination of elastic part) [34]. The crossover frequencies (ω c) for all the samples at 293–333 K are presented in Table 4. It is evident from Table 4 that the (G c) values will shift toward low-frequency region as both moduli increased. This may be due to enhanced entanglements present between the polymeric chains representing the intermolecular interaction strength which will slower the relaxation time (Fig. 6).

Representative plot of dynamic moduli as a function of angular frequency of MANIPAM-1 at 293 K
According to sticky reputation theory of Leibler, the characteristic time (t c) is the time at which the disentanglement of the polymeric chains takes place. It can be calculated using the following equation and its values are given in Table 4.
It has been observed that the relaxation time (t c) increases linearly with increasing concentration of polymer forming the stronger permanent junctions in the gel. It can be ascribed to increase entanglements in the structure due to larger chains. Similar observations have been observed for many other systems [35].
Conclusion
In summary, we synthesized chemically cross-linked methacrylamide-acrylic acid-N-isopropylacrylamide hydrogels. TGA revealed R 3, F 2/3 mechanism for the first stage of thermal degradation with E a = 36.9 kJ mol−1 and A = 3 × 106 s−1, while P1/4 for the second and third stages of polymer degradation with E a = 114.9, 217.7 kJ mol−1 and A = 2 × 1010, 4 × 1015 s−1, respectively. DSC study showed that the glass transition temperature of the polymeric samples increased with increasing methacrylamide contents in the polymeric sample indicating the increase in the extent of cross-linking. Flow curves verified the shear thinning behavior in all the synthesized samples at all the temperatures studied establishing their pseudo-plastic nature. Ostwald, modified Bingham, and Herschel–Bulkley models were well fitted to the rheological data for investigation of fluidity index and yield stress, respectively. Furthermore, relaxation time increased with temperature representing the stronger permanent junctions in the prepared samples of gel.
References
Stuart MAC et al (2010) Emerging applications of stimuli-responsive polymer materials. Nat Mat 9(2):101–113
Kumar A, Raj R, Kailas SV (2015) A novel in situ polymer derived nano ceramic MMC by friction stir processing. Mater Des 85:626–634
Singh S, Prasher S (2004) The etching and structural response of Makrofol-N and Makrofol-KG polycarbonate to γ-irradiation. Radiat Eff Defect Solids 159(6):359–367
Harris FW (1981) Introduction to polymer chemistry. J Chem Educ 58(11):837–843
Qiu Y, Park K (2001) Environment-sensitive hydrogels for drug delivery. Adv Drug Deliv Rev 53(3):321–339
Chen G, Hoffman AS (1995) Graft copolymers that exhibit temperature-induced phase transitions over a wide range of pH. Nature 373(6509):49–52
Seddiki N, Aliouche D (2013) Synthesis, rheological behavior and swelling properties of copolymer hydrogels based on poly (N-isopropylacrylamide) with hydrophilic monomers. Bull Chem Soc Ethiop 27(3):447–457
Shah N, Patel K (2014) Formulation and development of hydrogel for poly acrylamide-co-acrylic acid. J Pharm Sci Biosci Res 4:114–120
Ahamed MR et al (2010) Cation exchange properties of a terpolymer: synthesis and characterization. Int J Chem Environ Eng 1(1):7–12
Islam MT et al (2004) Rheological characterization of topical carbomer gels neutralized to different pH. Pharm Res 21(7):1192–1199
Akbar J et al (2012) A QSPR study of drug release from an arabinoxylan using ab initio optimization and neural networks. Carbohyd Polym 88(4):1348–1357
Silva R et al (2010) Miscibility influence in the thermal stability and kinetic parameters of poly (3-hydroxybutyrate)/poly (ethylene terephthalate) sulphonated blends. Polimeros 20(2):153–158
Rajeshwar K (1983) Thermal analysis of coals, oil shales and oil sands. Thermochim Acta 63(1):97–112
Jaber J, Probert S (2000) Non-isothermal thermogravimetry and decomposition kinetics of two Jordanian oil shales under different processing conditions. Fuel Process Technol 63(1):57–70
Akbar J et al (2012) Kinetics and mechanism of thermal degradation of pentose-and hexose-based carbohydrate polymers. Carbohyd Polym 90(3):1386–1393
Khawam A, Flanagan DR (2006) Solid-state kinetic models: basics and mathematical fundamentals. J Phys Chem B 110(35):17315–17328
Pienaar A, Vilakazi B (2012) Gas-phase fluorination kinetics of Ta2O5 with elemental fluorine. J South Afr Inst Min Metall 112:545–549
Turmanova SC et al (2008) Non-isothermal degradation kinetics of filled with rise husk ash polypropene composites. Express Polym Lett 2(2):133–146
Vlaev L et al (2008) A comparative study of non-isothermal kinetics of decomposition of calcium oxalate monohydrate. J Anal Appl Pyrol 81(2):253–262
Georgieva V, Zvezdova D, Vlaev L (2013) Non-isothermal kinetics of thermal degradation of chitin. J Therm Anal Calorim 111(1):763–771
Ruvolo-Filho A, Curti PS (2006) Chemical kinetic model and thermodynamic compensation effect of alkaline hydrolysis of waste poly (ethylene terephthalate) in nonaqueous ethylene glycol solution. Ind Eng Chem 45(24):7985–7996
Wissemborski R, Klein R (2010) Welding and marking of plastics with lasers. Laser Tech J 7(5):19–22
Guan J, Chen G (2010) Copolymerization modification of silk fabric with organophosphorous flame retardant. Fire. Mater 37(5):261–270
Dumitriu RP, Cazacu G, Vasilescu DS (2015) Rheological behavior of alginate-lignosulfonate blend solutions. In: Proceedings of the 5th European conference of chemical engineering (ECCE’15). Mathematical and computational methods in applied sciences, vol 15, pp 161–165
Al-Shammari B et al (2011) The effect of polymer concentration and temperature on the rheological behavior of metallocene linear low density polyethylene (mLLDPE) solutions. J KSU Eng Sci 23(1):9–14
Al-Fariss T, Al-Zahrani S (1993) Rheological behaviour of some dilute polymer solutions. Eng Sci 5(1):95–109
Alakali JS-A, Irtwange SV, Mkavga M (2009) Rheological characteristics of food gum (Cissus populnea). Afr J Food Sci 3(9):237–242
Mohammed MA, Mohammed AJ (2009) Effect of type and concentration of different water soluble polymer solutions on rheological properties. NUCEJ 12:26–37
Bird RB, Armstrong RC, Hassager O (1987) Dynamics of polymeric liquids. Vol. 1: Fluid mechanics. Wiley-Interscience, New York
Mastalska-Popławska J et al (2016) Rheology of cross-linked poly(sodium acrylate)/sodium silicate hydrogels. Arab J Sci Eng 41(6):2221–2228
Tonon R et al (2009) Steady and dynamic shear rheological properties of açai pulp. J Food Eng 92(4):425–431
Shchipunov YA, Hoffmann H (2000) Thinning and thickening effects induced by shearing in lecithin solutions of polymer-like micelles. Rheol Acta 39(6):542–553
Okamoto M et al (2001) A house of cards structure in polypropylene/clay nanocomposites under elongational flow. Nano Lett 1(6):295–298
Liu R et al (2014) Rheological properties of hydrophobically associative copolymers prepared in a mixed micellar method based on methacryloxyethyl-dimethyl cetyl ammonium chloride as surfmer. Int J Polym Sci 2014:1–14
Tae G et al (2001) Hydrogels with controlled, surface erosion characteristics from self-assembly of fluoroalkyl-ended poly (ethylene glycol). Macromol 34(18):6409–6419
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Kousar, F., Malana, M.A., Chughtai, A.H. et al. Synthesis and characterization of methacrylamide-acrylic acid-N-isopropylacrylamide polymeric hydrogel: degradation kinetics and rheological studies. Polym. Bull. 75, 1275–1298 (2018). https://doi.org/10.1007/s00289-017-2090-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-2090-3