
Abstract
Poly(1,4-butadiene)-graft-poly(L-lactide) copolymers were efficiently prepared via the grafting-from strategy using a combination of ring-opening metathesis polymerization (ROMP) of a hydroxyl-functionalized cyclobutene monomer and organocatalyzed ring-opening polymerization (ROP) of L-lactide. The copolymers were characterized by NMR, MALDI-TOF, SEC, TGA, and DSC demonstrating that well-defined polyester-grafted poly(1,4-butadiene) were obtained through the reported methodology.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Bottlebrush copolymers, which possess a very high density of polymer arms uniformly grafted to a backbone of a linear polymer, represent a unique class of polymers because of their interesting morphologies and properties [1]. The synthesis of densely grafted copolymers typically involves the use of one or more living/controlled polymerization methods [2]. Thanks to the orthogonality of ring-opening metathesis polymerization (ROMP) toward organocatalyzed ring-opening polymerization (ROP) and the highly active ruthenium-based olefin metathesis catalyst [3, 4], ROMP has emerged as an effective chemistry to reach narrowly dispersed graft copolymers based on polyenes and polyesters with high molar masses [5]. Indeed, numerous works have reported the synthesis of graft copolymers from “ROMP-able” (oxa)norbornene derivatives because of their high reactivity. Our group has developed a number of efficient synthetic strategies based on the cyclobutene ring as the “ROMP-able” functionality to prepare well-defined graft copolymers having a strictly poly(1,4-butadiene) (PBu) main chain with a high density of grafts [6]. These PBu-grafted copolymers have been synthesized using the grafting-from or the grafting-through approach [7,8,9,10]. Use of the grafting-from technique has allowed us to synthesize well-defined high molar mass PBu-g-poly(methyl methacrylate), PBu-g-poly(tert-butyl acrylate) and PBu-g-poly(ε-caprolactone) graft copolymers [9, 10].
In this work, we report the synthesis and characterization of bottlebrush PBu-g-poly(L-lactide) (PBu-g-PLLA) copolymers obtained from the grafting-from strategy. Besides distinct properties among other polyesters, polylactide has a unique behavior related to stereocomplex crystallization between its two stereoisomers, i.e., PLLA and PDLA [11, 12]. In the literature, the few previous reports devoted to the synthesis of lactide-grafted ROMP polymers using the grafting-from approach started from a polynorbornene [13, 14], polynorbornene-b-polyoxanorbornene [15] or poly(norbornene-stat-cyclooctadiene) backbone [16, 17]. Herein, we expand this methodology to the grafting of PLA. PLA arms were synthesized from a PBu backbone containing one pendant hydroxyl in every repeating unit used as the initiating group for the organocatalyzed ROP of L-lactide (L-LA) mediated by 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD). Thermal properties of the resulting PBu-g-PLLA graft copolymers are reported.
Experimental
Materials
All chemicals were purchased from Aldrich unless otherwise noted. Acetic acid (98.0%, Merck), chloroform (CHCl3, >99.8%), n-hexane (>97.0%), silica gel for chromatography (SiO2, 0.035–0.070 mm, 60 Å, Acros Organics) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD, 98%) were used as received. L-Lactide (L-LA, 98%) was recrystallized in toluene three times prior to use. Dichloromethane (DCM, HPLC grade, Fisher Chemical) and toluene (HPLC grade, Fisher Chemical) were dried over dry solvent stations GT S100. Poly(1,4-butadiene)-based ring-opening polymerization (ROP) macroinitiators containing a pendant hydroxyl in each repeating unit (PBu) were synthesized according to the previously reported procedure [10].
General characterization
NMR spectra were recorded on an Advance DPX 200 spectrometer for 1H NMR (200 MHz). Chemical shifts are reported in ppm relative to the deuterated solvent resonances. The average molar masses (Number-average molar mass \( \overline{{M_{n} }} \), weight-average molar mass \( \overline{{M_{w} }} \)) and dispersity (Ð = \( \overline{{M_{w} }} /\overline{{M_{n} }} \)) values were measured by Size Exclusion Chromatography (SEC) using tetrahydrofuran (THF) as the eluent and carried out using a system equipped with a SpectraSYSTEM AS 1000 autosampler, with a guard column (Polymer Laboratories, PL gel 5 µm Guard column, 50 × 7.5 mm) followed by two columns (Polymer Laboratories, 2 PL gel 5 µm MIXED-D columns, 2 × 300 × 7.5) and with a SpectraSYSTEM RI-150 detector. The instrument operated at a flow rate of 1.0 mL min−1 at 35 °C and was calibrated with narrow linear polystyrene (PS) standards ranging in molar mass from 580 to 483,000 g mol−1. Matrix-Assisted Laser Desorption and Ionization Time Of Flight (MALDI-TOF) mass spectrometry analysis was performed on a Bruker UltraFlex II MALDI-TOF instrument equipped with a nitrogen laser operating at 337 nm, a 2 GHz sampling rate digitizer, a pulsed ion extraction source and a reflectron. The laser pulse width is 3 ns and the maximum power is 200 mJ. Spectra were recorded in the linear mode with an acceleration voltage of 19 kV and a delay of 200 ns. 100 single shot acquisitions were summed to give the spectra and the data were analyzed using Bruker FlexAnalysis and Polytools softwares. Samples were prepared by dissolving the matrix (trans-2-[3-(4-tert-butylphenyl)-2-methyl-2-propenylidene]malononitrile, DCTB) in DCM (30 mg mL−1) and mixing with the polymer (2 mg mL−1) in the ratio 1:50 (v/v). An aliquot of this solution (1 μL) was spotted directly onto a thin layer of sodium trifluoroacetate in acetone (concentration 19 mg mL−1) that had been deposited to act as a cationizing agent. Thermogravimetric analyses (TGA) were performed on a TA Instruments Q500 apparatus measuring the total mass loss on approximately 10 mg samples from 30 °C up to 600 °C at a heating rate of 10 °C min−1 in a nitrogen flow of 90 mL min−1. Differential scanning calorimetry (DSC) measurements were performed on a TA Instruments Q100 connected to a computer in aluminum pans under nitrogen otherwise noted. Samples were heated from 20 to 100 °C, held at 100 °C for 20 min to erase any history effects, and then cooled to −80 °C, kept at −80 °C for 5 min, and heated again to 200 °C at a heating rate of 5 °C min−1. Crystallization temperatures (T c) were obtained from the cooling scan, while glass transition (T g) and melting transition temperatures (T m) were obtained from the second heating scan.
General procedure for ROP of L-LA from PBu
A dry 10 mL Schlenk flask equipped with a stirring bar, a vial, two rubber septums, PBu, L-LA and TBD were introduced into a glovebox. L-LA (0.500 g, 3.47 mmol) and the desired quantity of PBu were added into the Schlenk flask. The Schlenk flask was then capped with a rubber septum. TBD (20 mg, 144 μmol) was added into the vial and then capped with a rubber septum. The Schlenk flask and the vial were removed from the glovebox. 0.4 mL of degassed anhydrous DCM was then introduced via a syringe into the vial ([TBD] = 359 mmol L−1). 5 mL of degassed anhydrous DCM was then introduced via a syringe into the Schlenk flask and allowed to stir. When a homogeneous solution was obtained, the Schlenk flask was immersed in an oil bath preset at 25 °C and 100 μL of the TBD solution is added via a syringe to allow the polymerization to proceed (initial reaction time, t = 0). The polymerization was quenched after 1 h by the addition of acetic acid solution in toluene (0.2 mL, 16.5 mmol L−1). The resulting mixture was then concentrated to dryness under vacuum. The crude polymer was then dissolved in DCM (2 mL) and precipitated into n-hexane (50 mL). The crude PBu-g-PLLA copolymers were dissolved in CHCl3 and passed through a short SiO2 column (5 g for a 0.2 g PBu-g-PLLA sample) using CHCl3 as the eluting solvent. The resulting polymer solution volumes were reduced, and white copolymers were recovered by precipitating into n-hexane.
Poly(1,4-butadiene)-g-poly(L-lactide) PBu 85 - g - P L LA 15
White plastic. [L-LA]0/[OH]0/[TBD]0 = 35/1/0.35 (Table 1, run 2); \( \overline{{M_{{n,{\text{NMR}}}} }} \) = 201,044 g mol−1; \( \overline{{M_{{n,{\text{SEC}}}} }} \) = 194,600 g mol−1; Ð = 1.07. 1H NMR (200 MHz, CD3COCD3), δ (ppm): 7.25–7.10 (bs, 425H, CH2–C6 H 5), 5.20–4.95 (t, J = 7.4 Hz, 2550H, CH–CH3 of the PLLA repeating unit), 1.52–1.30 (d, J = 7.2 Hz, 7650H, CH–CH 3 of the PLLA repeating unit) (Fig. S3 in Supporting Information).
Results and discussion
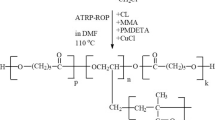
The grafting-from approach was used to synthesize poly(1,4-butadiene) (PBu) backbones with poly(L-lactide) (PLLA) grafts. Ring-opening polymerization (ROP) of L-lactide (L-LA) was performed following a procedure reported earlier [8] using PBu backbones containing pendant hydroxyl in every repeating unit as the ROP macroinitiator and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) as the catalyst (Scheme 1). TBD was chosen as the catalyst as it proved highly efficient in mediating the ROP of L-LA [18, 19].

Synthesis of poly(1,4-butadiene)-g-poly(L-lactide) graft copolymers according to the grafting-from strategy (Bn = CH2-Ph)
The ROP of L-LA was carried out from PBu-based ROP macroinitiators with a number-average degree of polymerization (\( \overline{{{\text{DP}}_{n} }} \)) of 85 and 198 containing pendant hydroxyl in every repeating unit with [L-LA]0/[OH]0 feed ratios ranging from 16/1 to 35/1 (Scheme 1; Table 1). The polymerization was performed at 25 °C in dichloromethane (DCM) for 1 h and then quenched by the addition of acetic acid. After precipitation in n-hexane, the crude copolymers were passed through a short column of silica to remove a small amount of linear PLLA homopolymer presumably formed by adventitious initiation [17] and transesterification side reactions [20] (backbiting), as evidenced by the SEC profile showing a lower molar mass population (see Fig. S1 in Supporting Information) and Matrix-Assisted Laser Desorption and Ionization Time Of Flight (MALDI-TOF) mass spectrometry analysis applied to the low molar mass population (see Fig. S2 in Supporting Information). These side reactions lead to a lack of agreement between the monomer-to-initiator ratio and the experimental number-average degree of polymerization (\( \overline{{{\text{DP}}_{n} }} \)) of the PLA grafts, determined from 1H NMR analysis of the purified PBu-g-PLLA copolymers (Table 1, run 2) by comparing the peak integration area of the methyl group of PLLA at 1.35–1.50 ppm (labeled (b) in Fig. S3 in Supporting Information) with those of aromatic protons of the benzyl group at δ = 7.10–7.45 ppm (labeled (c) in Fig. S3 in Supporting Information). Nevertheless, the experimental \( \overline{{{\text{DP}}_{n} }} \) values increased with increasing [L-LA]0/[OH]0 ratio (Table 1, run 1 vs 2). Furthermore, the clear shift of the SEC traces toward higher molar masses with increasing [L-LA]0/[OH]0 ratios and a narrow and unimodal elution peak with Ð in the range of 1.07–1.32 (Table 1; Fig. 1 and Fig. S4 in Supporting Information) indicate that the PLLA graft extension was effective to form well-defined PBu-g-PLLA copolymers. The differences between \( \overline{{M_{{n,{\text{NMR}}}} }} \) and \( \overline{{M_{{n,{\text{SEC}}}} }} \) is the result of the difference in hydrodynamic volumes of the graft copolymers compared to linear polystyrene standards used to calibrate the SEC [7].

DSC analysis was used to determine the thermal properties of PBu-g-PLLA graft copolymers and those of linear PLLA homopolymer [8] (Fig. 2). The T g of linear PLLA homopolymer of similar \( \overline{{M_{n} }} \) as the PLLA grafts in the PBu-g-PLLA copolymer was observed at 37 °C (see Fig. 2b). This value is close to those reported for linear PLLA homopolymers with similar \( \overline{{M_{n} }} \) and functionalized by norbornene or hepta-1,6-diyne chain end [21, 22]. The melting (T m) and crystallization (T c) temperatures of linear PLLA are observed at 126 and 87 °C, respectively (see Fig. 2b), together with the well-known double-melting behavior of PLLA [23]. The crystallinity of the samples, calculated by comparison with the reported enthalpy of fusion for the parent polymer crystal (∆H f 100% cristalline PLLA = 93 J g−1) [24], was 37%, and is similar with values reported in the literature for different linear PLLA homopolymers [21, 22, 25]. DSC trace of PBu 85 - g -P L LA 15 indicates a T g at 41 °C but did not reveal any T m (see Fig. 2a). A similar observation was reported in the literature [22, 25] for graft copolymers with short side chains of PLLA ≤ 35. This behavior can be ascribed to the confinement of the grafts that inhibits their mobility and capability for crystallization [22, 25].

DSC curves for (a) PBu 85 - g -P L LA 15 (full line) (Table 1, run 2), (b) P L LA 18 (dotted dashed line) and (c) PBu 85 (dashed line)
The thermal decomposition profiles of copolymer PBu 85 - g -P L LA 15 (Fig. 3c) and of the corresponding polymer precursors (Fig. 3a, b) were characterized via thermogravimetric analysis (TGA). The TGA curve of the PBu 85 - g -P L LA 15 copolymer showed a two-step thermal decomposition: the first step can be caused by the decomposition of PLLA in the range from 225 to 300 °C, and the second step is attributed to the decomposition of the PBu backbone. A similar observation was reported in the literature for polynorbornene-g-poly(D,L-lactide) copolymers [26]. This broad degradation temperature range for polylactide is a signature of the random chain scission accompanied by transesterification reactions [26].

TGA curves for (a) PBu 85 (dashed line), (b) P L LA 18 (dotted dashed line), and (c) PBu 85 - g -P L LA 15 (full line) (Table 1, run 2)
Conclusion
In summary, PBu backbones bearing one pendant hydroxyl in every repeating unit have been successfully used as macroinitiators for the organocatalyzed ROP of L-LA by TBD to prepare original PBu-g-PLLA copolymers through the grafting-from route. These polymers were thoroughly characterized by NMR, MALDI-TOF, SEC, TGA, and DSC to reveal their structure, sizes, molar masses, and thermal properties. The results are expected to be useful in the design and synthesis of complex hollowed nanostructures by taking advantage of the hydrolytic (bio)degradation PLLA side chains of PBu-g-PLLA.
References
Hadjichristidis N, Pitsikalis M, Iatrou H, Driva P, Chatzichristidi M, Sakellariou G, Lohse D (2014) Graft copolymers. In: Mark HM (ed) Encyclopedia of polymer science and technology, 4th edn. Wiley, New York, pp 467–526
Hadjichristidis N, Iatrou H, Pitsikalis M, Mays J (2006) Macromolecular architectures by living and controlled/living polymerizations. Progr Polym Sci 31:1068–1132
Grubbs RH (2006) Olefin-metathesis catalysts for the preparation of molecules and materials (nobel lecture). Angew Chem Int Ed 45:3760–3765
Grubbs RH, Khosravi E (2015) Polymer synthesis. Handbook of metathesis, vol 3, 2nd edn. Wiley-VCH, Weinheim
Knall AC, Slugovc C (2014) Olefin metathesis polymerization. In: Grela K (ed) Olefin metathesis. Wiley, New York, pp 269–284
Le D, Morandi G, Legoupy S, Pascual S, Montembault V, Fontaine L (2013) Cyclobutenyl macromonomers: synthetic strategies and ring-opening metathesis polymerization. Eur Polym J 49:972–983
Morandi G, Montembault V, Pascual S, Legoupy S, Fontaine L (2006) Well-defined graft copolymers issued from cyclobutenyl macromonomers by combination of ATRP and ROMP. Macromolecules 39:2732–2735
Leroux F, Montembault V, Pascual S, Guerin W, Guillaume SM, Fontaine L (2014) Synthesis and polymerization of cyclobutenyl-functionalized polylactide and polycaprolactone: a consecutive ROP/ROMP route to poly(1,4-butadiene)-g-polyesters. Polym Chem 5:3476–3486
Morandi G, Pascual S, Montembault V, Legoupy S, Delorme N, Fontaine L (2009) Synthesis of brush copolymers based on a poly(1,4-butadiene) backbone via the “grafting from” approach by ROMP and ATRP. Macromolecules 42:6927–6931
Leroux F, Montembault V, Piogé S, Pascual S, Brotons G, Fontaine L (2016) High molar mass poly(1,4-butadiene)-graft-poly(ε-caprolactone) copolymers by ROMP: synthesis via the grafting-from route and self-assembling properties. Macromolecules 49:4739–4745
Pensec S, Leroy M, Akkouche H, Spassky N (2000) Stereocomplex formation in enantiomeric diblock and triblock copolymers of poly(ε-caprolactone) and polylactide. Polym Bull 45:373–380
Tsuji H (2005) Poly(lactide) stereocomplexes: formation, structure, properties, degradation, and applications. Macromol Biosci 5:569–597
Jha S, Dutta S, Bowden NB (2004) Synthesis of ultralarge molecular weight bottlebrush polymers using Grubbs’ catalysts. Macromolecules 37:4365–4374
Zhang K, Tew GN (2012) Cyclic brush polymers by combining ring-expansion metathesis polymerization and the “grafting from” technique. ACS Macro Lett 1:574–579
Yoo J, Runge MB, Bowden NB (2011) Synthesis of complex architectures of comb block copolymers. Polymer 52:2499–2504
Jing F, Hillmyer MA (2008) A bifunctional monomer derived from lactide for toughening polylactide. J Am Chem Soc 130:13826–13827
Theryo G, Jing F, Pitet LM, Hillmyer MA (2010) Tough polylactide graft copolymers. Macromolecules 43:7394–7397
Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, Nederberg F, Choi J, Wade C, Waymouth RM, Hedrick JL (2006) Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters. Macromolecules 39:8574–8583
Pratt RC, Lohmeijer BGG, Long DA, Waymouth RM, Hedrick JL (2006) Triazabicyclodecene: a simple bifunctional organocatalyst for acyl transfer and ring-opening polymerization of cyclic esters. J Am Chem Soc 128:4556–4557
Penczek S, Szymanski R, Duda A, Baran J (2003) Living polymerization of cyclic esters—a route to (bio)degradable polymers. Influence of chain transfer to polymer on livingness. Macromol Symp 201:261–269
Li A, Li Z, Zhang S, Sun G, Policarpio DM, Wooley KL (2012) Synthesis and direct visualization of dumbbell-shaped molecular brushes. ACS Macro Lett 1:241–245
Kang EH, Lee IH, Choi TL (2012) Brush polymers containing semiconducting polyene backbones: graft-through synthesis via cyclopolymerization and conformational analysis on the coil-to-rod transition. ACS Macro Lett 1:1098–1102
Yasuniwa M, Tsubakihara S, Sugimoto Y, Nakafuku C (2004) Thermal analysis of the double-melting behavior of poly(L-lactic acid). J Polym Sci Part B: Polym Phys 42:25–32
Pyda M, Bopp RC, Wunderlich B (2004) Heat capacity of poly(lactic acid). J Chem Thermodyn 36:731–742
Sveinbjörnsson BR, Miyake GM, El-Batta A, Grubbs RH (2014) Stereocomplex formation of densely grafted brush polymers. ACS Macro Lett 3:26–29
Oh S, Lee JK, Theato P, Char K (2008) Nanoporous thin films based on polylactide-grafted norbornene copolymers. Chem Mater 20:6974–6984
Acknowledgements
We thank Mireille Barthe and Alexandre Bénard for SEC analyses, Emmanuelle Mebold for MALDI-TOF analyses, and Amélie Durand and Corentin Jacquemmoz for NMR analyses.
Author information
Authors and Affiliations
Corresponding authors
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Leroux, F., Montembault, V., Piogé, S. et al. Poly(1,4-butadiene)-graft-poly(L-lactide) via the grafting-from strategy. Polym. Bull. 74, 4415–4422 (2017). https://doi.org/10.1007/s00289-017-1961-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-017-1961-y