
Abstract
Au/poly[anthranilic acid-co-3-carboxy-N-(2-thenylidene)aniline/PVAc] [Au/P(ANA-co-CNTA)/PVAc] electrospun nanofibers were fabricated in different electrospinning media including acetone and dimethylformamide (DMF) for covalent immobilization of glucose oxidase (GOx). The surface of copolymer nanofibers was activated by EDC/NHS chemistry, and the presence of Au nanoparticles as tiny conduction centers inside the copolymer matrix enhanced the electrochemical properties. Morphology and composition of enzyme-immobilized nanofibers were characterized by scanning electron microscopy/energy-dispersive X-ray spectroscopy (EM/EDX) and atomic force microscope (AFM). The effective covalent binding of glucose oxidase onto the Au/P(ANA-co-CNTA)/PVAc nanofibers was also confirmed by FTIR-ATR and Raman spectroscopy. EIS measurements revealed that the charge transfer resistances of the enzyme-immobilized nanofibers were decreased with increasing amount of enzyme. The effect of electrospun nanofiber diameter on sensing properties of enzyme-functionalized nanofibers was investigated by EIS. The sensitivities of electrodes calculated from impedance measurement were 7.24 × 106 and 6.67 × 103 Ω mM−1 cm−2 for the Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOX (acetone), respectively. The impedance measurement results revealed that the linear range of Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) was lower than Au/P(ANA-co-CNTA)/PVAc-GOX (acetone). It could be attributed that smaller fiber diameter resulted in the higher specific surface area. This contributes to increasing the number of available active sites and, thus, increasing the amount of the enzyme loading.
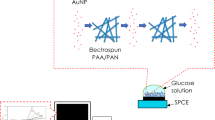
Graphical abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Enzymes are efficient catalysts with excellent properties including highly selective activity and specificity that accelerate the rate of chemical reactions without being consumed in the process [1].
However, the using of enzymes in the industrial applications due to their instability and non-reusability, is a challenging process. From such a viewpoint, covalent immobilization of enzymes by the surface functional groups of substrate as binding reaction sites has been recognized as an attractive strategy. Covalent immobilization has numerous advantages including enhanced and stable enzyme activity, facilitation of substrate, better product recovery, easier prevention of product contamination and reducing the costs of industrial applications [2]. Enzyme 3D structure is essential to its activity, then selecting support matrix and designing the carrier are very important in enzyme immobilization process.
Recently, conducting polymers in the forms of one-dimensional nanostructured (fibers) have attracted much attention as an enzyme immobilization support due to their highly porous structure, large surface area, and smaller size of the fiber, high loading, higher conductivity and easy and low-cost preparation [3]. Among various conducting polymers, polyaniline nanofibers (PANFs) provide an ideal support for the immobilization of the enzyme owing to their suitability as matrices for the immobilization and entrapment of enzymes [4]. However, the low process ability of polyaniline nanofibers, due to their low solubility, limits their applications. An effective way to overcome these limitations is using polyaniline derivatives such as poly anthranilic acid (PANA). It is preferred due to its solubility in aqueous and non-aqueous solvents as well as other polar solvents. Also, PANA nanofibers by containing carboxyl groups (–COOH) on their main structure have shown enormous potential for enzyme immobilization since amide linkages are formed by the condensation reaction of the carboxyl groups with amino groups of the enzymes [5, 6]. However, fabrication of PANA nanofibers with electrospinning is a challenging process due to their rigid backbones and relatively low molecular weight forms. To overcome this problem, blending of PANA with another electrospinnable polymer like polyvinyl acetate (PVAc) is needed since it offers good adhesion to most surfaces and can be dissolved in many solvents other than water [7].
In the past decades, Au nanoparticles with large surface area and good electronic properties have attracted continuous interest in many applications including, drug delivery [8], radiotherapy [9], cancer diagnosis [10], sensors and biosensors [11] and protein delivery systems and biological sensors [8]. Au nanoparticles act as conduction centers in the nanofiber structure to facilitate and improve electron transfer between the redox center of the enzyme and provide a stable host matrix for enzyme immobilization [12–14]. So, combination of both PANA with carboxyl groups and gold nanoparticles as tiny electrical centers in the fiber shape has exhibited excellent properties of both conducting polymer and nanoparticles as substrate for enzyme immobilization [15].
Monitoring of blood glucose level in biomedical applications is of great significance due to the metabolic disorder resulting from insulin deficiency and hyperglycemia when glucose concentrations in empty stomach are higher than the normal range of about 3.9–6.2 mM (2 h after eating something 3.9–7.8 mM). It can reduce the risk of diabetes mellitus due to induced heart disease, blindness and kidney failure by rapid and early detection of glucose levels in the blood [16–18]. Covalent immobilization of GOx as sensing biomolecule into the nanofiber mat, for the determination of glucose concentration, has attracted enormous attention.
Up to now, many attempts have been made for monitoring the glucose level. Different techniques used for detection of glucose level, including near-infrared spectrometry [19], surface plasmon resonance [20], fluorescence [21], colorimetry [22], and electrochemistry [23]. Electrochemical biosensors based on biomolecules immobilized on an electrode surface are promising tools due to their good selectivity, high sensitivity, easy and low-cost operation and compatibility for miniaturization [24]. In our recently published articles, gold/poly(anthranilic acid) (Au/PANA) core/shell nanoparticles were synthesized by in situ polymerization [25], and their electrospun nanofibers were fabricated by blending with polyvinyl acetate (PVAc) and were utilized as a stable support with high electrochemical properties for successful protein immobilization [26]. In this study, we report the nanofiber formation of core/shell [Au/P(ANA-co-CNTA)] nanostructures which was previously performed by us [27] where oxidation of the monomers and reduction of Au(III) take place simultaneously by forming composite structures (metal–polymer) without using any other initiators by incorporation of poly 3-carboxy-N-(2-thenylidene)aniline (CNTA) into the Au/PANA core/shells. Thereafter, in this work, Au/P(ANA-co-CNTA)/PVAc nanofibers were fabricated in two different solvent medium (DMF and acetone) for GOx immobilization to observe the effect of solvent on fiber diameter and sensing performance. Incorporation of gold nanoparticles as efficient electron transfer centers in the copolymer structure containing (COOH) groups as active sites enhanced the electrocatalytic activity toward H2O2 which was releasing enzymatic reaction of glucose. Glucose oxidase (GOx) is a well-known and commercially available enzyme for potential biosensor [13]. Since 1-ethyl-3-(dimethylaminopropyl) carbodiimide hydrochloride (EDC)/N-hydroxyl succinimide (NHS) activation process for covalent immobilization of enzyme has been widely applied to various kinds of substrates of polymers [5], Au/P(ANA-co-CNTA)/PVAc nanofibers were bioactivated by EDC/NHS chemistry for covalent immobilization of enzyme using (–COOH) groups of copolymer, especially PANA-modified gold nanoparticles and amine groups on the enzyme (Fig. 2). Surface morphologies of enzyme-functionalized nanofibers were analyzed by SEM/EDX and AFM. The interaction between the enzyme and the carboxylic acid groups was investigated by EIS analysis and equivalent circuit modeling. The obtained data revealed the effective immobilization of enzyme on the nanofiber surface. The sensing of glucose with fabricated electrodes was investigated by the electrochemical impedance spectroscopy.
Experimental section
Reagents and materials
Polyvinyl acetate (PVAc; molecular weight 500.00 g/mol), 3-aminobenzoic acid (anthranilic acid) and 3-carboxy-N-(2-thenylidene)aniline were purchased from Sigma-Aldrich. Glucose oxidase from Aspergillus niger (GOX, 228,253 U/g) was used in the form of lyophilized powder as received. N-(3-Dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride (EDC) and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich. Dimethylformamide (DMF) and acetone were supplied from Merck. Phosphate buffer solution (PBS, 0.05 M and pH 6.5) was prepared freshly before each experiment. All chemicals used in the experiments were of analytical reagent grade. Double-distilled deionized water was used in all of the experiments.
In this method, the initiator is HAuCl4, and the oxidation of the monomers and reduction of metal ions Au(III) to Au(0) take place simultaneously for forming composite structures (metal–polymer) (Fig. 1). The molar ratio of the anthranilic acid (ANA) to HAuCl4 is 1:1 (0.041 M), and the initial concentration of 3-carboxy-N-(2-thenylidene)aniline is 0.0024 M. The synthesis of Au/P(ANA-co-CNTA) nanocomposites was carried out according to our previous work [27]. In details, 200 mg sodium dodecyl sulfate (SDS) was added to 100 ml of doubly distilled water inside the three-neck round-bottomed flask equipped with stirrer to form micelles. After stirring for 5 h, 3-aminobenzoic acid (0.57 g) was added to the flask and stirred in an ultrasonic bath for 30 min. After complete dissolution of this monomer, 3-carboxy-N-(2-thenylidene)aniline (0.057 g) was added to the solution and stirred under the same conditions in ultrasonic bath. After the dissolution of the monomers, HAuCl4 (2.87 ml) was added dropwise to the solution, and polymerization was initiated and carried out at 28 °C for about 24 h. Finally, the precipitated brown polymer was collected by filtration, washed and dried in vacuum at 80 °C for 2 days.

Schematic representation of the reaction mechanism of P(ANA-co-CNTA) formation
Preparation of Au/P(ANA-co-CNTA)/PVAc blends
0.01 g of Au/P(ANA-co-CNTA) nanocomposite was dispersed in % 10 wt polyvinyl acetate (PVAc) prepared in DMF and acetone solvents, separately, and both the solutions were stirred for 24 h overnight to obtain a homogeneous solution for optimum electrospinning.
Fabrication of Au/P(ANA-co-CNTA)/PVAc nanofibers
An electrospinning device with 1 ml/h feed rate powered by a syringe pump (ES50 model, Gamma High Voltage Inc., USA) was used for the Au/P(ANA-co-CNTA)/PVAc electrospun nanofibers preparation. The electrospinning solutions were placed into a 2-ml syringe with a 0.7-mm blunt tip needle. The Au/P(ANA-co-CNTA)/PVAc electrospun nanofibers were directly deposited onto ITO-PET (NV Innovative Sputtering Technology, Zulte, Belgium, PET 175 μm, Coating ITO-60) substrate with dimensions of 0.5 × 2.5 cm, under the following electrospinning conditions: applied voltage 15 kV, spinning time 5 min, and working distance 15 cm.
Immobilization of GOX on Au/P(ANA-co-CNTA)/PVAc nanofibers
First, to activate COOH groups on the copolymer structure, fabricated Au/P(ANA-co-CNTA)/PVAc nanofibers were treated by EDC/NHS mixture which contained 0.015 M EDC and 0.03 M NHS prepared in phosphate buffer solution (PBS, pH 6.5, 0.05 M), and the nanofibers were treated by EDC/NHS solution with constant shaking (200 rpm) for 3 h at room temperature to obtain maximum activation. Activated nanofibers were then washed with PBS solution (0.05 M, pH 6.5) to remove excess EDC/NHS amount. Thereafter, nanofibers were immersed in glucose oxidase solution dissolved in phosphate buffer with concentration of 5 mg/ml. Nanofiber mats were incubated with glucose oxidase with constant shaking (200 rpm) for 3 h at +4 °C. Finally, enzyme-immobilized nanofibers were taken out, washed again with phosphate buffer solution two times at 4 °C to remove remaining non-covalent immobilized enzyme and stored at 4 °C for characterization. Schematic representation of the activation of Au/P(ANA-co-CNTA)/PVAc nanofibers by EDC/NHS chemistry for GOx immobilization is shown in Fig. 2.

Schematic representation of the activation of Au/P(ANA-co-CNTA)/PVAc nanofibers by EDC/NHS chemistry for GOx immobilization
Characterization of nanofibers before and after GOx immobilization
The morphology of nanofibers was investigated with SEM/EDX (QUANTA 400 F). Raman spectroscopy was performed using Raman analyzer (Thermo, DXR Raman), using a diode laser of wavelength 780 nm as excitation source with a power of 14 mW at a spectral resolution of 5 cm−1, and a CCD detector. Raman spectra were recorded in the range between 200 and 3300 cm−1. FTIR spectrum was recorded by FTIR-ATR spectrophotometer (Perkin Elmer, Spectrum One, with a universal ATR attachment with a diamond and a ZnSe crystal). Structural characterization of Au/P(ANA-co-CNTA)/PVAc nanofibers was investigated with tunneling electron microscopy (TEM) (JEOL JEM 2100). The electrocatalytic activity of the electrodes was evaluated in phosphate buffer solution (PBS 0.1 M) by cyclic voltammetry (CV) and electrochemical impedance spectroscopy with frequency range (0.01 Hz–10 kHz) and AC voltage of 10 mV using Ivium vertex (software, Ivium soft and Faraday cage, BASI Cell Stand C3). Electrochemical impedance spectroscopy was also performed by electrochemical analyzer (Princeton Applied Research, USA) with frequency range (0.01 Hz–100 kHz) and AC voltage of 10 mV. Three-electrode system, consisting of nanofiber mat as working electrode, platinum wire as counter electrode, and silver wire as pseudo-reference electrode, was used. All measurements were repeated three times for confirmation. The measured impedance spectra were analyzed in terms of electrical equivalent circuits using the analysis program ZSimpWin V.3.10.
Au/P(ANA-co-CNTA)/PVAc electrodes (DMF and acetone)
The electrochemical biosensing of glucose was performed under the optimized experimental conditions. Cyclic voltammetry measurements were performed on Au/P(ANA-co-CNTA)/PVAc electrodes prepared in DMF and acetone solvent medium in phosphate buffer solution (PBS, pH 6.5, 0.1 M) at a potential scan rate of 50 mVs−1 containing glucose concentrations from 20 µM to 0.1 mM and 3–10 mM, respectively. EIS measurements were performed on Au/P(ANA-co-CNTA)/PVAc electrodes prepared in DMF and acetone solvent medium in PBS (0.1 M and pH 6.5) containing glucose concentrations from 20 µM to 0.11 mM and 2–11 mM with frequency range between 0.01 Hz and 10 kHz with a perturbation amplitude of 10 mV at −0.4 V DC potential, respectively. To determine the linear working range of the electrodes, charge transfer resistance (R CT) as a function of glucose concentration was obtained by analyzing the EIS data in terms of electrical equivalent circuit of R(QR).
Results and discussions
TEM and EDX characterization of Au/P(ANA-co-CNTA)/PVAc nanofibers
The structure of Au/P(ANA-co-CNTA)/PVAc nanofibers was investigated with transmission electron microscopy (TEM) analysis, and the TEM image of Au/P(ANA-co-CNTA)/PVAc nanofiber is shown in Fig. 3, which confirms the distribution of gold nanoparticles in the nanofiber structure. As can be seen, the gold nanoparticles, with higher density, were much darker than the polymeric matrix due to the density variation of gold nanoparticles and polymer matrix [28].

TEM image of Au/P(ANA-co-CNTA)/PVAc nanofibers
Energy-dispersive X-ray spectroscopy (EDX) was used to determine the amount of gold and sulfur atoms in the Au/P(ANA-co-CNTA)/PVAc nanofibers, and also the distribution of the gold nanoparticles in the nanofibers was monitored by EDX mapping and represented in Fig. 4. As can be seen, the amount of gold atoms in the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) was 0.23 and 0.27 %, respectively (Table 1). Also, the amount of sulfur atoms in the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) was 0.03 and 0.04 %, respectively.

EDX mapping of Au/P(ANA-co-CNTA)/PVAc (DMF) (a) and Au/P(ANA-co-CNTA)/PVAc (acetone) (b)
FTIR-ATR characterization of Au/P(ANA-co-CNTA)/PVAc nanofibers
FTIR-ATR spectrophotometry was carried out to confirm the incorporation of poly 3-carboxy-N-(2-thenylidene)aniline in the Au/P(ANA-co-CNTA)/PVAc nanofibers structure. Significant difference between the spectra of Au/PANA/PVAc and Au/P(ANA-co-CNTA)/PVAc and also Au/P(ANA-co-CNTA)/PVAc and 3-carboxy-N-(2-thenylidene)aniline monomer is seen in Fig. 5. The strong stretching vibration of carboxyl group (C=O) of PANA was at around 1728 cm−1 at Au/PANA/PVAc (Fig. 5), however, it was shifted to 1730 cm−1 in the case of Au/P(ANA-co-CNTA)/PVAc nanofibers [25]. The peak at 1220 cm−1, generally assigned to the C–N+• stretching vibration in the polaron structure, indicates that the PANA is in the doped state [29]. The bands centered at 1360 and 1545 cm−1 are originated to symmetric and asymmetric C=C stretching vibrations of the thiophene ring. Also, S–H stretching of thiophene ring appeared at 2356 cm−1 [30]. The peak at 878 cm−1, also assigned to the C–S bending band, provides incorporation of 3-carboxy-N-(2-thenylidene) aniline into the final copolymeric nanofibers.

FTIR spectra of Au/P(ANA-co-CNTA)/PVAc and [Au/PANA/PVAc nanofibers and 3-carboxy-N-(2-thenylidene)aniline (inset)]
FTIR-ATR and Raman characterization of GOX-immobilized nanofibers
FTIR-ATR spectra of Au/P(ANA-co-CNTA)/PVAc nanofibers were obtained before and after glucose oxidase immobilization to confirm the covalent immobilization of glucose oxidase onto the nanofibers (Fig. 6a, b). The appearance of sharp absorption bands at 1655 and 1647 cm−1 in the FTIR spectra of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) nanofibers is attributed to the formation of amide bond after immobilization of GOx, respectively [31]. In the FTIR spectrum of GOx-immobilized nanofibers (DMF and acetone), sharp peaks at 2369 and 2357 cm−1 were assigned to R–N+ group at Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx (acetone), respectively, which is due to the protonated form (imidazolium) of histidine side chain [32]. These peaks were overlapped with S–H stretching peak of thiophene ring at 2356 cm−1, and the peak absorbance of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF and acetone) nanofibers increased in comparison with bare Au/P(ANA-co-CNTA)/PVAc nanofibers which confirms the immobilization of glucose oxidase onto the membrane (inset of Fig. 6a, b) [32]. The obvious peaks at 3300 and 3289 cm−1 in the spectra of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) belong to the O–H stretching of hydroxyl groups of the GOx molecule, respectively [31]. The results were in agreement where GOx really exists in the electrospun nanofibers. The assignments of the FTIR-ATR bands of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF and acetone) are listed in Table 2.

FTIR spectra of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) (a), Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) (b) before and after immobilization of glucose oxidase and Raman spectra of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) (c) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) (d) before and after immobilization of glucose oxidase
Raman spectra of the GOx, Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF and acetone) are shown in Fig. 6c, d. Presence of peaks at 1615 and 1621 cm−1 was attributed to the C–C stretching of benzoic stretching at Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), respectively [33]. The bands that centered at about 1300 and 1293 cm−1 were assigned to the C–N+• stretching of PANA at Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), respectively [34].The peaks at 860 and 850 cm−1 are attributed to the amine deformation of benzenoid ring of PANA at Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), respectively [35]. The characteristic bands of the C=C ring stretching of thiophene rings in the thiophene unit chains appeared at 1468 and 1462 cm−1 at Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), respectively [36]. Two peaks at 1195 and 1183 cm−1 are attributed to the C–H bending in benzenoid structures of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and (acetone), respectively [33]. The bands at (709, 704 cm−1) and 1097 cm−1 and (1004, 1000 cm−1) belong to C–S–C ring deformation, C–H bending and C–S stretching of thiophene units at Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), respectively [36, 37]. These results confirmed the presence of thiophene units in the matrix and formation of copolymer. By comparing the Raman spectra of the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) (Fig. 6c, d) and GOx (inset of Fig. 5) with those of the enzyme-immobilized electrodes, a new peak appeared at 1415 cm−1 which is a characteristic peak of the CO2 − symmetric stretching vibration in amino acid which is seen at 1440 cm−1 for GOx-immobilized nanofibers (inset of Fig. 6c, d) indicating that GOx is immobilized onto the nanofibers’ surface [38]. The assignments of the Raman bands of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF and acetone) are listed in Table 3.
Morphological and structural investigation of Au/P(ANA-co-CNTA)/PVAc electrodes (DMF and acetone) before and after GOx immobilization (SEM and AFM)
The morphology of Au/P(ANA-co-CNTA)/PVAc nanofibers under both conditions, before and after glucose oxidase immobilization, was investigated with SEM. The average diameters of nanofibers were determined using ImageJ by randomly measuring the diameters of ten individual fibers shown in SEM images. The average diameters of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF), before and after immobilization of glucose oxidase, were 440 ± 178 nm and 550 ± 190 nm, respectively (Fig. 7a, b). However, the average fiber diameter of Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) was 1200 ± 750 nm which increased to 3220 ± 1721 nm after immobilization of enzyme (Fig. 7c, d). The increase in the nanofibers’ diameter indicates the successful attachment of the glucose oxidase onto the nanofibers.

SEM images of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) (a), Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF) (b), Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) (c) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (acetone) (d)
To investigate the morphology of the Au/P(ANA-co-CNTA)/PVAc nanofibers before and after enzyme immobilization, AFM was used. Figure 8a–f shows the AFM images of the immobilized and non-immobilized nanofibers. Au/P(ANA-co-CNTA)/PVAc nanofibers prepared in DMF and acetone solution have RMS roughness of 45.46 and 70.00 nm, respectively. However, the surface roughness was slightly increased after glucose oxidase immobilization. The RMS roughness values of Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (acetone) were 66.84 and 82.59 nm, respectively. This increase in roughness indicates the successful immobilization of GOx on the surface of activated Au/P(ANA-co-CNTA)/PVAc nanofibers [39].

AFM images of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) (a), Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF) in 8 μm (b) and 20 μm magnification (c), Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) (d), Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (acetone) in 8 μm (e) and 20 μm magnification (f)
Electrochemical impedance spectroscopy (EIS) and equivalent circuit modeling
Electrochemical impedance spectroscopy (EIS) has been a popular tool to monitor the electrode assembly process due to its immediate and sensitive response to the surface changes [40]. Electrical properties of the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) on ITO-PET were studied before and after GOx immobilization by electrochemical impedance spectroscopy (EIS). Nyquist (Figs. 9a, 10a) and Bode phase (Figs. 9b, 10b) plots of the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) and Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) were recorded by applying an AC signal of 10 mV amplitude in the frequency range 0.01 Hz–100 kHz, to confirm the presence of GOx on the surface of nanofibers. Significant differences in the Nyquist and Bode phase were observed depending on the covalent immobilization of glucose oxidase onto the nanofibers. Bare Au/P(ANA-co-CNTA)/PVAc nanofibers showed a linear behavior with a higher slope in Nyquist plots, indicating the capacitive behavior of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone), while enzyme-immobilized Au/P(ANA-co-CNTA)/PVAc-GOx nanofibers (DMF and acetone) showed a linear line with a depressed semicircle at Nyquist plots (Figs. 9a, 10a).

Nyquist (a) and Bode phase (b) plots obtained after GOx immobilization onto the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) indicating that measured and calculated data well fitted to each other with the model

Nyquist (a) and Bode phase (b) plots obtained after GOx immobilization onto the Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) indicating that measured and calculated data well fitted to each other with the model
Figures 9b and 10b represent Bode phase plots of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone), and the values were ~85°. After GOx immobilization onto the Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone), from intermediate to high frequencies, there was no significant change in phase angle values, indicating that diffusion process was slightly dominated through nanofibers’ changes in Bode phase plot [41] at medium to low frequency domain; changes in phase angle values were observed and the phase angle values were decreased (around 65) which is due to the presence of immobilized enzyme as additional capacitive elements onto the nanofibers [42, 43].
The EIS spectra were analyzed in terms of electrical equivalent circuits modeled to evaluate the kinetics of the systems using ZSimpWin software, and the calculated data and measured data were well fitted with the chosen equivalent circuit (Fig. 11). The impedance spectra of Au/P(ANA-co-CNT)/PVAc (DMF and acetone) and enzyme-immobilized Au/P(ANA-co-CNT)/PVAc nanofibers (DMF and acetone) are described by the equivalent circuit of R(QR) (Fig. 11) in short hand. Table 4 represents the fitting parameters for the equivalent circuit elements by modeling of the impedance spectra of nanofibers after GOX immobilization.

Equivalent circuit for the simulation of the EIS spectra of Au/P(ANA-co-CNTA)/PVAc nanofibers before and after GOx immobilization
The first component represents the solution resistance of the electrolyte (Rs) due to the presence of the electrolyte on the nanofiber in the solution [13] or maybe of the electrical contacts [44, 45].
According to Table 4, the Rs values of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) were dramatically increased after GOx immobilization. These data correlated with the data obtained from AFM images. Since, after GOx immobilization, surface roughness increased significantly, then it is expected to have higher solution resistance in comparison to the bare Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone), and also due to the presence of high enzyme immobilized onto the bare Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) in comparison with Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone), the Rs is increasing more significantly.
The second resistance in both circuits represents the charge transfer resistance (Rct) between the electrolyte and the surface of the Au/P(ANA-co-CNTA)/PVAc nanofibers or enzyme-immobilized nanofibers (DMF and acetone) [43].
The charge transfer resistances (Rct) of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF) and Au/P(ANA-co-CNTA)/PVAc nanofibers (acetone) were 2.4E8 and 15.7 Ω, respectively, however, after GOx immobilization, the charge transfer resistance values decreased to 1.04 and 0.65 Ω, respectively. Since all the EIS measurements were performed in PBS buffer solution with a pH 6.5, the GOx molecule is negatively charged, because the isoelectric point (Ip) of GOx lies at pH 4.2 [46].
Positively charged ions can interact with negatively charged GOx on the surface, and negatively charged ions interact with PANA on the electrode surface, and solvated counter ions align along the nanofiber surface [43, 47]. Therefore, charge transfer resistance of Au/P(ANA-co-CNTA)/PVAc (DMF and acetone) nanofibers is decreased after immobilization of GOx.
Q corresponds to the constant phase element (CPE) that was associated with non-ideal behavior due to the surface roughness and anomalous transport effects and the electrode resulting from the nanofiber composition [47, 48]. The formula [Z CPE = T CPE (jw)−n] is used to define the impedance of a non-ideal electrode. T CPE and n are frequency-independent constants, w is the angular frequency, n is a parameter describing the deviation from an ideal capacitor and arises from the slope of the log Z versus log f plot [25]. The values for n vary from 0 to 1. n = 1 subscribes to an ideal capacitor, while n = 0 and 0.5 denote resistance and Warburg behavior, respectively [43]. The n values of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone) were around 0.94, indicating that they behave like ideal capacitors. But after enzyme immobilization, the n values of Au/P(ANA-co-CNTA)/PVAc nanofibers (DMF and acetone) were slightly decreased to 0.83 and 0.89, respectively, which indicate more resistive behavior compared to nanofibers before enzyme immobilization.
Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co CNTA)/PVAc-GOx (acetone) as glucose sensors
GOx exhibits a catalytic activity toward glucose oxidation by dissolving oxygen. Glucose can be used for the detection of the enzymatic release of H2O2 by electrochemical method. GOx catalyzes the oxidation of glucose in the presence of O2 to produce gluconic acid and generates hydrogen peroxide (H2O2) [49]. Thus, H2O2 could be determined by electrochemical technique such as CV, chronoamperometry, and EIS.
The cyclic voltammograms of the Au/P(ANA-co-CNTA)/PVAc-GOx electrodes (DMF and acetone) recorded using various concentrations of glucose (20 µM–0.1 mM) for Au/P(ANA-co-CNTA)/PVAc-GOx (DMF), and 3–10 mM for Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) electrodes in 0.1 M PBS (pH 6.5) electrolyte at a scan rate of 50 mVs−1 are shown in Fig. 12a, b. The redox peaks were observed relating to the reduction of H2O2 and probably O2 in the voltammograms [44, 50, 51]. The reduction peak current both in Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) and Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) electrodes increased linearly with increasing concentration of glucose. This indicated that GOx-immobilized Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) electrodes exhibit electro-activity toward the reduction of H2O2 [43]. Figure 12c, d represents the variation of peak current response with glucose concentration for both the Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc–GOx (acetone) electrodes, respectively. The calibration curve of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVA-GOx (acetone) corresponding to amperometric response (Fig. 12c and inset of Fig. 12d) was linear against the concentrations of glucose ranging from 20 µM to 0.1 mM for Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) (R 2 = 0.9906) and 3–7 mM for Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) (R 2 = 0.9866), respectively. The sensitivities of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) were calculated as 20.3733 and 0.3952 µA mM−1 cm−2, respectively. The linear range of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) was lower than Au/P(ANA-co-CNTA)/PVAc-GOx (acetone). It could be attributed that the smaller fiber diameter and the higher specific surface area contribute to the increase in the number of available active sites, thus increasing the amount of enzyme loading.

Cyclic voltammogram of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) electrode in PBS solution containing various concentrations of glucose at scan rate of 50 mVs−1 (a), Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) electrode in PBS solution containing various concentrations of glucose at scan rate of 50 mVs−1 (b), calibration curve of Au/P(ANA-co-CNTA)/PVAc-GOx (DMF) current density vs. glucose concentration (mM) (c), and current density vs. glucose concentration for Au/P(ANA-co-CNTA)/PVAc-GOx (acetone); inset shows corresponding calibration of Au/P(ANA-co-CNTA)/PVAc-GOx (acetone) (mM) (d)
Variation of glucose concentration was also investigated by EIS measurements of the Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOX (acetone) on the successive additions of glucose to 0.1 M PBS (pH 6.5).
H2O2 which is transformed by GOX can be determined by the charge transfer reaction [52]. Figure 13a, b shows the Nyquist plots of electrodes in the increasing concentrations of glucose. The linear correlation (Fig. 13c, d) for glucose was obtained by plotting charge transfer resistance (R CTn − R CTbare) obtained from the equivalent circuit modeling R(QR) as a function of glucose concentration. The R CT of Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) (Fig. 13c) increased linearly with the concentration of glucose in the linear range of 30 µM–0.11 mM according to linear regression equation: y = 6.41 × 105 + 7.24 × 106 x with a squared correlation coefficient, R 2, of 0.9424. The sensitivity of Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) electrode was 7.24 × 106 Ω mM−1 cm−2. Au/P(ANA-co-CNTA)/PVAc-GOX (acetone) electrode (Fig. 13d) displayed a linear response for glucose concentrations ranging from 3 to 7 mM, according to the linear equation: y = 6.18 × 104 + 6.67 × 103 x, with a squared correlation coefficient, R 2, of 0.9866. The sensitivity of electrode was determined as 6.67 × 103 Ω mM−1 cm−2. The calibration curves obtained from impedance measurements considerably exhibited the same linear range as that obtained from amperometric response. Impedance measurements also revealed that the Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) electrode had a low linear range than Au/P(ANA-co-CNTA)/PVAc-GOX (acetone) electrode.

Nyquist plots of Au/P(ANA-co-CNTA)/PVAc-GOX nanofibers (DMF) (a) and Au/P(ANA-co-CNTA)/PVAc-GOX nanofibers (acetone) in 0.1 M PBS (pH 6.5) buffer solution at different glucose concentrations; frequency range between 0.01 Hz and 10 kHz with a perturbation amplitude of 10 mV DC potential of −0.4 V(b), calibration curve of Au/P(ANA-co-CNTA)/PVAc-GOX(DMF) RCTn–RCTBar e vs. glucose concentration (mM) (c) and charge transfer resistance (RCTn–RCTBare) vs. glucose concentration for Au/P(ANA-co-CNTA)/PVAc-GOx (acetone); inset shows calibration curve of Au/P(ANA-co-CNTA)/PVAc-GOX (acetone) RCTn–RCTBare vs. glucose concentration (mM) (d)
Conclusion
Glucose oxidase (GOx) was successfully immobilized via covalent bonding onto the Au/P(ANA-co-CNTA)/PVAc nanofibers with different fiber diameters. The effect of fiber diameters on enzyme immobilization, and also morphological, structural and electrochemical characterization of enzyme-immobilized nanofibers were studied by SEM/EDAX, AFM, FTIR-ATR, Raman, CV and EIS. The sensitivities of Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) and Au/P(ANA-co-CNTA)/PVAc-GOX (acetone) electrodes were calculated as 20.3733 and 0.3952 µA mM−1 cm−2 from cyclic voltammetric measurements, and 7.24 × 106 and 6.67 × 103 Ω mM−1 cm−2 from impedimetric measurements, respectively. The impedance and amperometric measurement results revealed that the linear range of Au/P(ANA-co-CNTA)/PVAc-GOX (DMF) was lower than Au/P(ANA-co-CNTA)/PVAc-GOX (acetone). It could be attributed that smaller fiber diameter results in the higher specific surface area. This contributes to increasing the number of available active sites, thus increasing the amount of the enzyme loading.
Abbreviations
- TEM:
-
Transmission electron microscopy
- SEM:
-
Scanning electron microscopy
- AFM:
-
Atomic force microscopy
- EIS:
-
Electrochemical impedance spectroscopy
- FTIR-ATR:
-
Fourier transform infrared spectroscopy
- P(ANA-co-CNTA):
-
Poly(anthranilic acid-co-3-carboxy-N-(2-thenylidene)aniline)
- PVAc:
-
Poly(vinyl acetate)
- EDX:
-
Energy-dispersive X-ray spectroscopy
- EDC:
-
(1-Ethyl-3-(3-dimethylamino)propyl) carbodiimide
- NHS:
-
N-Hydroxysuccinimide
- PANA:
-
Poly anthranilic acid
- SDS:
-
Sodium dodecyl sulfate
- GOx:
-
Glucose oxidase
- PBS:
-
Phosphate buffer solution
- RMS:
-
Root mean square
- DC:
-
Direct current
- ITO-PET:
-
Indium tin oxide-polyethylene terephthalate
References
Koeller KM, Wong CH (2001) Enzymes for chemical synthesis. Nature 409:232–240
Min D, Zhang X, He W et al (2013) Direct immobilization of glucose oxidase in magnetic mesoporous bioactive glasses. J Mater Chem B 1:3295
Arecchi A, Scampicchio M, Brenna OV, Mannino S (2010) Biocatalytic nylon nanofibrous membranes. Anal Bioanal Chem 398:3097–3103
Kima H, Leea I, Kwonb Y et al (2011) Immobilization of glucose oxidase into polyaniline nanofiber matrix for biofuel cell applications. J Biosens Bioelectron 26:3908–3913
Sarac AS, Guler Z (2016) Electrochemical impedance and spectroscopy study of the EDC/NHS activation of the carboxyl groups on poly(ε-caprolactone)/poly(m-anthranilic acid) nanofibers. Express Polym Lett 10:2
Giray D, Balkan T, Dietzel B, Sarac AS (2013) Electrochemical impedance study on nanofibers of poly(m-anthranilic acid)/polyacrylonitrile blends. Eur Polym J. 49:2645–2653
Verma SK, Bisarya SC (1986) Improvement in properties of poly(vinyl acetate): emulsion with dibasic acids. J Appl Polym Sci 31:2675–2684
Thomas M, Klibanov A (2003) Conjugation to gold nanoparticles enhances polyethylenimines transfer of plasmid DNA into mammalian cells. Proc Natl Acad Sci USA 100:9138–9143
Hirsch LR, Stafford RJ, Bankson JA (2003) Nanoshell-mediated nearinfrared thermal therapy of tumors under magnetic resonance guidance. Proc Natl Acad Sci USA 100:13549–13554
Cai W, Gao T, Hong H, Sun J (2008) Applications of gold nanoparticles in cancer nanotechnology. Nanotechnol Sci Appl. 1:17–32
Du D, Chen S, Cai J, Zhang A (2007) Immobilization of acetylcholinesterase on gold nanoparticles embedded in sol-gel film for amperometric detection of organophosphorous insecticid. Biosens Bioelectron 23:130–134
Petkova GA, Záruba K, Žvátora P, Král V (2012) Gold and silver nanoparticles for biomolecule immobilization and enzymatic catalysis. Nanoscale Res Lett 7:287
Hecht HJ, Kalisz HM, Hendle J et al (1993) Crystal structure of glucose oxidase from Aspergillus niger. J Mol Biol. 229:153
Conde J, Ambrosone A, Sanz V et al (2012) Design of multifunctional gold nanoparticles for in vitro and in vivo gene silencing. ACS Nano 6:8316–8324
Shan Ye J, Sheu F-S (2007) Carbon nanotube-based sensor. In: Kumar CSSR (ed) Nanomaterials for biosensors. Wiley-VCH, Weinheim, pp 27–89
Heller A, Feldmen B (2008) Electrochemical glucose sensors and their applications in diabetes management. Chem Rev 108:2482–2505
Chen C, Xie Q, Yang D et al (2013) Recent advances in electrochemical glucose biosensors: a review. RSC Adv 3:4473–4491. doi:10.1039/C2RA22351A
Shukla SK, Mishra AK, Mamba BB, Arotiba OA (2013) Amperometric and photometric responses of in situ coupled glucose oxidase-poly (propylene imine) dendrimer based glucose biosensor. Int J Electrochem Sci 8:11711–11722
Mesch M, Zhang C, Braun PV, Giessen H (2015) Functionalized hydrogel on plasmonic nanoantennas for noninvasive glucose sensing. ACS Photonics 2:475–480
He H, Xu X, Wu H et al (2013) In situ nanoplasmonic probing of enzymatic activity of monolayer-confined glucose oxidase on colloidal nanoparticles. Anal Chem 85:4546–4553
Lan MH, Zhang JF, Chui YS et al (2015) Carbon nanoparticles based ratiometric fluorescent sensor for detecting mercury ions in aqueous media and living cells. J Mater Chem B 3:127–134
Xia Y, Ye J, Tan K et al (2013) Colorimetric visualization of glucose at the submicromole level in serum by a homogenous silver nanoprism–glucose oxidase system. Anal Chem 85:6241–6247
Gao X, Wang N, Jia S, Shao Y (2013) Detection of glucose based on bimetallic PtCu nanochains modified electrodes. Anal Chem 85:5040–5046
Grieshaber D, MacKenzie R, Voro\({{\ddot{\text s}}}\) J, Reimhult E (2008) Electrochemical biosensors—sensor principles and architectures. Sensors 8:1400–1458
Golshaei R, Guler Z, Ünsal C, Sarac AS (2015) In situ spectroscopic and electrochemical impedance study of gold/poly (anthranilic acid) core/shell nanoparticles. Eur Polym J 66:502–512
Golshaei R, Guler Z, Sarac SA (2016) (Au/PANA/PVAc) nanofibers as a novel composite matrix for albumin and streptavidin immobilization. Mater Sci Eng C 60:260–275
Golshaei R, Ghoreishi SM, Sarac AS (2015) Core–shell nanostructures of gold-copolymer of 3-carboxy-N-(2-thenylidene) aniline and anthranilic acid. Int J Nanoparticles Nanotech 1:002
Maleki M, Latifi M, Amani-Tehran M, Mathur S (2009) Electrospun core-shell nanofibers for drug encapsulation and sustained release. Polym Eng Sci 53:1770–1779
Zhang L, Peng H, Fang C et al (2007) Self-assembled polyaniline nanotubes grown from a polymeric acid solution. Nanotechnology 18:115607
Gnanakan SRP, Rajasekhar M, Subramania A (2009) Synthesis of polythiophene nanoparticles by surfactant-assisted dilute polymerization method for high performance redo supercapacitors. Int J Electrochem Sci 4:1289–1301
Hood AR, Saurakhiya N, Deva D, Sharma A, Verma N (2013) Development of bimetal-grown multi-scale carbon micro-nanofibers as an immobilizing matrix for enzymes in biosensor applications. Mater Sci Eng C 33:4313–4322
Rauf S, Ihsan A, Akhtar K et al (2006) Glucose oxidase immobilization on a novel cellulose acetate-polymethylmethacrylate membrane. J Biotechnol 121:351–360
Bachhav SG, Patil DR (2015) Synthesis and characterization of polyaniline-multiwalled carbon nanotube nanocomposites and its electrical percolation behavior. Am J Mater Sci 5:90–95
Berkkan A, Seçkin AI, Pekmez K, Tamer U (2010) Amperometric enzyme electrode for glucose determination based on poly(pyrrole-2-aminobenzoic acid). J Solid State Electrochem 14:975–980
Falcou A, Duchene A, Hourquebie P et al (2005) A new chemical polymerization process for substituted anilines: application to the synthesis of poly (N-alkylanilines) and poly (o-alkylanilines) and comparison of their respective properties. Synth Met 149:115–122
Tran-Van F, Garreau S, Louarn G et al (2001) Fully undoped and soluble oligo(3,4-ethylenedioxythiophene)s: spectroscopic study and electrochemical characterization. J Mater Chem B 11:1378–1382
Fu M, Chen F, Zhang J, Shi G (2002) Electrochemical fabrication of aligned microtubular heterojunctions of poly (p-phenylene) and polythiophene. J Mater Chem 12:2331–2333
Yang Y, Mu SH, Chen H (1998) Electrochemical synthesis of polypyrrole for the immobilization of galactose oxidase. Synth Met 92:173–178
Cho H-J, Perikamana SKM, Lee J-H et al (2014) Effective immobilization of BMP-2 mediated by polydopamine coating on bio-degradable nanofibers for enhanced in vivo bone formation. ACS Appl Mater Interfaces. 6:11225–11235
Yang WR, Gooding JJ, Hibbert DB (2001) Characterisation of gold electrodes modified with self-assembled monolayers of l-cysteine for the adsorptive stripping analysis of copper. J Electroanal Chem 516:10–16
Randles JEB (1947) Kinetics of rapid electrode reactions. Discuss Faraday Soc 1:11–19
De Crombrugghe A, Yunus S, Bertrand P (2008) Grafting and characterization of protein on polyaniline surface for biosensor applications. Surf Interface Anal 40(3–4):404–407
Dagli U, Guler Z, Sarac AS (2015) Covalent immobilization of tyrosinase on electrospun polyacrylonitrile/polyurethane/poly (m-anthranilic acid) nanofibers: an electrochemical impedance study. Polym Plast Technol Eng 54(2015):1494–1504
Anusha JR, Hee-JeKim T, Fleming A et al (2014) Simple fabrication of ZnO/Pt/chitosan electrode for enzymatic glucosecose biosensor. Sens Actuator B Chem 202:827–833
Zehani N, Dzyadevych SV, Kherrat R, Jaffrezic-Renault NJ (2014) Sensitive impedimetric biosensor for direct detection of diazinon based on lipases. Front Chem 2:1–7
Yan X, Xu XK, Ji H-F (2005) Glucose oxidase multilayer modified microcantilevers for glucose measurement. Anal Chem 77:6197–6204
Guler Z, Erkoc P, Sarac AS (2015) Electrochemical impedance spectroscopic study of single-stranded DNA-immobilized electroactive polypyrrole-coated electrospun poly(ε-caprolactone) nanofibers. Mater Express 5:269–279
Sadkowski A (2000) On the ideal polarisability of electrodes displaying cpe-type capacitance dispersion. J Electroanal Chem 481:222–226
Wu J, Yin F (2013) Sensitive enzymatic glucose biosensor fabricated by electrospinning composite nanofibers and electrodepositing Prussian blue film. J Electroanal Chem 694:1–5
Shan CH, Yang H, Song J et al (2009) Direct electrochemistry of glucosecose oxidase and biosensing for glucosecose based on graphene. Anal Chem 81:2378–2382
Palanisamy S, Ezhil Vilian AT, Chen S-M (2012) Direct electrochemistry of glucosecose oxidase at reduced graphene oxide/zinc oxide composite modified electrode for glucosecose sensor. Int J Electrochem Sci 7:2153–2163
Zane D, Appetecchi GB, Bianchini C et al (2011) An impedimetric glucose biosensor based on overoxidized polypyrrole thin film. Electroanalysis 23:1134–1141
Acknowledgments
The authors express their thanks to Scientific and Technological Research Council of Turkey (TUBITAK) for the financial support on Project 2216. We would like to thank Zeliha Guler for her valuable discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Golshaei, R., Karazehir, T., Ghoreishi, S.M. et al. Glucose oxidase immobilization onto Au/poly[anthranilic acid-co-3-carboxy-N-(2-thenylidene)aniline]/PVAc electrospun nanofibers. Polym. Bull. 74, 1493–1517 (2017). https://doi.org/10.1007/s00289-016-1786-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-016-1786-0