
Abstract
Monomer pairs, dimethyl N,N-diallylaspartate hydrochloride [(CH2=CH–CH2)2NH+CH(CO2Me)CH2CO2Me Cl−] (I)/SO2 and N,N-diallylaspartic acid hydrochloride [(CH2=CH-CH2)2NH+CH(CO2H)CH2CO2H Cl−] (II)/SO2, underwent alternate cyclocopolymerization to give cationic polyelecyrolytes (CPEs), poly(I-alt-SO2) (III) and poly(II-alt-SO2) (IV), respectively, in very good yields. (+) III upon acid hydrolysis was converted to cationic (+) IV, bearing in each repeating unit the triprotic acid residues [(NH+…(CO2H)2] of aspartic acid hydrochloride. Under the influence of pH, (+) IV has been equilibrated to water-insoluble diprotic polyzwitterionic acid (±) V, water-soluble monoprotic poly(zwitterion-anion) (± −) VI and finally its conjugate base polydianion (=) VII. Basicity constants of CO2 − and amine group have were determined. The pH-responsive polymer was demonstrated to be an efficient antiscalant against CaSO4 scaling. For potential separation and purification of biomolecules, a recyclable aqueous two-phase system (ATPS) was constructed using water-soluble (=) VII and urethanized polyvinyl alcohol; the ionic polymer can be recycled by precipitating it as (±) V at a lower pH.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Aspartic acid (1) is one of the 23 proteinogenic amino acids, the building blocks of proteins; it plays an important role in the citric acid cycle, or Krebs cycle, during which many other important amino acids are synthesized (Scheme 1) [1]. Aspartic acid also plays a crucial role in generating cellular energy and, as such, gets its reputation as a treatment for chronic fatigue. Poly(aspartic acid) (PASA) is a biodegradable water-soluble poly(amino acid) which may replace many non-biodegradable polymers [2]. Even though PASA has been found in nature as fragments of larger proteins [3], to our knowledge it is yet to be isolated as a pure homo polymeric material from any natural source [4]. Polyaspartate (PASP), a 30:70 mixture of α- and β-linked amino acid (3 and 4), is currently produced on the industrial scale by the thermal polymerization process, through the polysuccinimide (2) intermediate (Scheme 1) [5]. PASP and its derivatives are biodegradable [6] and environmentally friendly; as such, they are attractive alternatives to polyacrylic acid as corrosion inhibitors [5], antiscalant in desalination processes [7] and chelator of metal ions in wastewater treatment. They have also found applications in manufacturing super-swelling material in diaper products and food packaging [8], biodegradable detergent and dispersant [9]. Recent research has also paved the way for the use of PASA-based pH-sensitive hydrogel for controlled drug release [10].

Aspartic acid, polyaspartate and polymer containing aspartic acid residues
Note that in the peptide bonds in PASP, the nitrogen loses its basic character while each repeating unit consists of a single anionic center in the pendant carboxylate motifs. Keeping in view the broad interest on this material by the biomedical and material research community, diallyl derivative 5 of aspartic acid was cyclopolymerized using Butler’s cyclopolymerization protocol [11–14] to obtain low molecular weight polymer 6, in which the basic character of the nitrogen and the two anionic centers in the carboxylates are preserved [15]. However, attempts to obtain homopolymer 6 having high molar masses were unsuccessful. It has been reported earlier that diallylammonium salts having pendant carboxyl functionality give low molecular weight homopolymers in lower yields even in the presence of excessive amount of initiators [16]. No rationale was provided to account for the difficulty associated with the presence of the carboxyl moiety. The objective of the current work is to apply the copolymerization protocol [16] to obtain 5/SO2 copolymer 12 (Scheme 2), which would allow us to study the effect of SO2 spacer on its pH-responsive solubility behavior, basicity constant of the metal chelation centers and antiscalant properties. We intend to exploit the pH-dependent solubility behavior of 12 to develop recyclable aqueous two-phase system (ATPS) for potential application in bioseparation technology [17, 18]. The titled alternate copolymer would represent the first example of its kind, containing the residues of an important amino acid, i.e., aspartic acid. The current work would pave the way to utilize Butler’s cyclopolymerization protocol [19] to synthesize aspartic acid-based cross-linked adsorbents for scavenging toxic metal ions from wastewater.

Cyclopolymerization protocol in the synthesis of pH-responsive polymers containing aspartic acid residues
Experimental methods
Materials
For dialysis, membrane (Spectra/Por) with a MWCO of 6000–8000 was purchased from Spectrum Laboratories, Inc. (Rancho Dominguez, CA, USA). 2,2′-Azoisobutyronitrile (AIBN) from Fluka AG (Buchs, Switzerland) was crystallized from C2H5OH/CHCl3 mixture. Dimethylsulfoxide (DMSO) was dried over calcium hydride overnight and then distilled under reduced pressure at a boiling point of 64–65 °C (4 mmHg).
Polyvinyl alcohol (PVA) 16 with a \(\overline{M}_{\text{n}}\) of 72,000 g mol−1 and a degree of hydrolyzation of 0.975–0.995 mol fraction was purchased from Fluka Chemie AG (Buchs, Switzerland). Partly urethanized PVA (UPVA) 17 (urethanized to the extent of 0.13 mol fraction) of \(\overline{M}_{\text{n}}\) of 69,000 g mol−1 was synthesized by heating a PVA 16/urea in 1:1 mol ratio as described (Scheme 2) [20, 21]. Dimethyl N,N-diallylaspartate (9) upon treatment with dry HCl gave hydrochloride salt 10, while its hydrolysis with NaOH followed by acidification with HCl afforded N,N-diallylaspartic acid hydrochloride 5 [15].
Physical methods
Perkin Elmer Series II (Model 2400) was used to carry out elemental analysis, and IR spectra were recorded on a Perkin Elmer 16F PC FTIR spectrometer. The NMR spectra were taken in a JEOL LA 500 MHz spectrometer using the residual proton resonance of D2O at δ 4.65 ppm as an internal standard and dioxane 13C peak at δ 67.4 ppm as an external standard. Thermogravimetric analysis (TGA) was carried out using an SDT analyzer (Q600: TA instruments, USA) in a nitrogen atmosphere. Viscosities were measured in a Ubbelohde viscometer (viscometer constant = 0.005317 mm2/s2) using CO2-free water under N2 at 30.0 ± 0.1 °C. The pH of the solutions was measured by a Sartorius pH meter PB 11. Conductivity measurements were carried out using an Orion Versa Star benchtop meter Thermoscientific (Beverly, MA, USA). PL-GPC 220 from Agilent Technologies equipped with a refractive index (RI) detector and two mixed bed columns (PL-aquagel-OH mixed-H 8 µm, 300 × 7.5 mm) was used to determine the molecular weight. A 50 µl portion of a 0.100 w/v % aqueous solution of the polymer was injected into the GPC columns, and HPLC-grade water containing 0.003 M NaN3 was used as eluent at a flow rate of 1.0 mL/min.
Procedure for 10/SO2 copolymerization and physical characterization of 11
Table 1 describes the polymerization conditions. For instance, for the experiment under entry 4, SO2 (2.56 g, 40 mmol) was absorbed in a monomer 10 (11.1 g, 40 mmol)/DMSO (12 g) solution in an RB flask (50 mL). The initiator AIBN (0.120 g) was added and the mixture was stirred under N2 at 63 °C for 36 h. The transparent reaction mixture was precipitated in acetone. The white polymer was ground using mortar and pestle, filtered and washed with hot acetone. Copolymer 11 was then dried for 4 h at 60 °C under vacuum to a constant weight (11.8 g, 86 %). Mp. (closed capillary): slight phase change was noticed around 170 °C; it became darker on further heating and turned brown at 285 °C and black at 325 °C with no further changes up to 400 °C. (Found: C, 41.9; H, 6.1; N, 4.0; S, 9.2. C12H20NO6SCl requires C, 42.17; H, 5.90; N, 4.10; S, 9.38 %); v max (KBr): 3438, 2962, 2920, 1745, 1635, 1446, 1376, 1307, 1227, 1126, 1052, 1002, 857, 775 and 514 cm−1. The 1H and 13C NMR spectra are displayed in Figs. 1 and 2.

1H NMR spectrum of (a) 10, (b) 5, (c) 11, (d) 13 (in the presence of KI) and (e) top layer (System 1, Table 5) in D2O

13C NMR spectrum of (a) 10, (b) 5, (c) 11 and (d) 13 (in the presence of KI) in D2O
Acidic hydrolysis of copolymer 11
A solution of copolymer 11 (entry 4, Table 1) (10.3 g, 30 mmol) in 6 M HCl (120 cm3) was stirred at 65 °C for 72 h to complete the hydrolysis of the ester groups as checked by the absence of the ethoxy proton signals. During the hydrolysis, polymer gradually precipitated from the initially clear solution. The entire mixture was dialyzed against deionized water for 48 h to remove HCl. The white polymer was ground using a mortar and pestle, filtered, washed with liberal excess of water and dried under vacuum at 55 °C to obtain copolymer 13 (7.4 g, 89 %) as a white powder. Its spectral data are identical to that of the polymer obtained via 5/SO2 copolymerization as described below.
Monomer 5/SO2 copolymerization
The conditions for the copolymerization is described in Table 2. For instance, SO2 (1.28 g, 20 mmol) was absorbed in a solution of monomer 5 (5.0 g, 20 mmol) in DMSO (5.0 g) in an RB flask (25 mL). Initiator AIBN (0.100 g) was then added under N2 to the homogeneous solution; the mixture in the RB flask was stirred at 63 °C for 36 h. The immovable gel was soaked in water for 24 h and the white polymer was ground with mortar and pestle, filtered and washed with excess water to obtain copolymer 13, which was dried under vacuum at 60 °C to a constant weight. Its spectral data are identical to those of the polymer obtained via hydrolysis of the diester polymer 11 (vide supra). The thermal decomposition: turned tan on heating, became dark brown at 290 °C, and darker on further heating up to 400 °C with evolution of gas at 330 °C. (Found: C, 43.0; H, 5.6; N, 4.9, S, 11.2. C10H15NO6S requires C, 43.32; H, 5.45; N, 5.05; S 11.56 %); v max. (KBr): 3459, 3041, 2977, 2920, 1728, 1628, 1386, 1305, 1127, 862, 780, 669 and 512 cm−1. The 1H, 13C NMR spectra and TGA curve are displayed in Figs. 1, 2 and 3a, respectively.

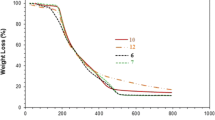
a TGA curve of PZA 13; using an Ubbelohde viscometer at 30 °C, the viscosity behavior of b (+) CPE 11 (entry 4, Table 1) in (i) filled square salt-free water, (ii) filled circle 0.1 M HCl and (iii) open square 0.1 M NaCl; c (±) PZA 13 (entry 1, Table 2) in (i) filled square 1 equivs NaOH, salt-free water, (ii) open square 2 equiv NaOH, salt-free water; (iii) filled circle 2 equivs NaOH, 0.1 M NaCl, (iv) open circle 1 equivs NaOH, 0.1 M NaCl, (v) filled triangle PZA 13 (prepared from entry 4, Table 1) 1 equiv NaOH, 0.1 M NaCl
Solubility measurements
The critical (minimum) salt concentration (CSC) values were determined by titrating a 1 % w/w aqueous solution of PZA 13 containing salts at a higher concentration than their CSC values at 23 °C with deionized water. The average of the triplicate results of the CSCs were determined to be 4.73 M NaBr and 2.90 M NaI with approximate accuracies of ± 2–3 %. The polymer was found to be insoluble in the presence of any concentration of NaCl or HCl (0–12 M).
Potentiometric titrations
Protonation constants (K) were determined using potentiometric titration as described elsewhere [22]. In each trial, a certain mmol of PZA 13 (\({\text{ZH}}_{2}^{ \pm }\)) was dissolved in a known amount of 0.1106 M NaOH (2.50 equivalent), then diluted to 200 cm3 using CO2-free water (Table 3). After each addition of 0.05–0.15 cm3 of 0.1222 M HCl, the recorded pH values were used to calculate the log K i s by the Henderson–Hasselbalch Eq. (2) (Scheme 2 contains Eqs. 1–3). Because of the solubility problem of PZA 13, log K 3 associated with the equilibration 13 (\({\text{ZH}}_{2}^{ \pm }\)) + H+ ⇋ (\({\text{ZH}}_{3}^{ + }\)) 12 could not be determined in salt-free water.
Evaluation of antiscalant behavior
Newly synthesized antiscalant 13 (dissolved using 1 equiv. NaHCO3, Table 2, entry 1) (20 and 30 ppm) was used to study the inhibition of calcium sulfate scale formation at 40 ± 1 °C in a supersaturated solution of 2598 mg/L of Ca2+ and 6300 mg/L of SO4 2− [23]. The induction time, defined as the time at which a rapid decrease in conductivity occurs, indicates the beginning of CaSO4 precipitation (Table 4).
Phase compositions of PZA 13 (+2 equivs NaOH)—UPVE 17–H2O (NaCl) systems
The tie lines by 1H NMR spectroscopy
The stock solutions (20 wt%) of PZA 13 (Table 2, entry 1: treated with 2.0 equivalents of NaOH) and UPVE 17 in 0.6 M NaCl were used in certain compositions to make several ATPSs of total volume ≈7 cm3 in a calibrated cylinder using diluents of 0.6 M NaCl. After mixing and centrifuging (ca. 5 min), the separated layers were equilibrated at 23 °C for 24 h. Volumes of the top and bottom layers were recorded and their densities measured. The 1H NMR of the phases after exchanging H2O with D2O were taken to determine the molar ratios of the polymers in each phase. Figure 1e displays the NMR spectrum of the top phase of system 1 (Table 5); the 13 protons of the PZA 13 (+NaOH) appeared as NMR signals in the range 2.3 ≤ δ ≥ 3.7 ppm with an area integration of A, while the two-proton signals for UPVA 17 is displayed around δ 1.7 ppm with an area of B. Integration of the signals thus provided the mole ratios of the polymers 13/17 in each phase as (A/13)/(B/2). The molar mass of the repeat unit of PZA 13 and UPVA 17 are taken as 277.3 and 49.6 g mol−1, respectively. After determining the weight fraction (w) in each phase as described [20], the tie lines were constructed.
Binodals by turbidity method
The binodal data for the 13–17-water (NaCl) systems at 23 °C were obtained by turbidimetric method using a procedure described elsewhere [20].
Results and discussions
Synthesis of polymers and their characterization
Dimethyl N,N-diallyl aspartate (9), the Michael addition product from diallyamine (7)-dimethyl maleate (8), on treatment with gaseous HCl gave cationic monomer 10, which upon hydrolysis with NaOH followed by acidification afforded the hydrochloride salt of N,N -diallyl aspartic acid (5) (Scheme 2).
Monomers 10 and 5 underwent cyclocopolymerization in the presence of initiator AIBN to give cationic polyelectrolyte (CPE) 11 (Table 1) and polyzwitterionic acid (PZA) 13 (Table 2), respectively, in very good to excellent yields. The change in initiator concentration from 3 mg/mmol monomer (Table 1, entry 1) to 4 and 5 mg/mmol (entries 2 and 3) did not change either the yield or the intrinsic viscosity in 0.1 M HCl. However, the presence of higher amount of solvent DMSO (0.30 g/mmol monomer in entry 4 vs. 0.25 g/mmol monomers in entries 1–3) afforded the polymer in the highest yield (Table 1, entry 4). The amount of initiator also did not have any considerable effect on the yield and viscosity values of PZA 13 (Table 2). The polymers in Tables 1 and 2 were correlated by hydrolyzing CPE 11 (from entry 4) to CPE 12, which upon dialysis was converted into PZA 13 upon depletion of HCl (Scheme 2).
Three major weight losses were revealed in the TGA curve of PZA 13 (Fig. 3a). The first slow weight loss of 8 % up to 100 °C resulted from the release of moisture trapped in the polymer. The polymer remained stable in the temperature range 100–150 °C; thereafter, a second sharp loss of 23 % in the temperature range 150–300 °C resulted from the release of SO2. The third steep loss of 40 % in the temperature range 300–430 °C was associated with the decarboxylation and the degradation of the nitrogenated organic fraction. At 800 °C, the residual mass was found to be 17 %.
Attempts to analyze the molar masses of PZA 13 by GPC failed due to strong adsorption to the column materials. A similar difficulty associated with polymers containing amine and carboxy motifs has been reported [24]. However, CPE 11 from entries 3 and 4 (Table 1) were determined to have \(\overline{M}_{W}\) of 58,500 and 56,000 g mol−1, respectively, with a PDI of 2.2.
Solubility behavior
CPE 11 as well as poly(zwitterion-anion) (PZAN) (± −) 14 and polydianionic electrolyte (PDE) (=) 15 are water soluble, as expected of any polyelectrolyte or polymer backbone with a charge imbalance in favor of either of the algebraic signs. It is the anionic portion in (± −) 14 that promotes water solubility. It is worth mentioning that in 0.1 M NaCl, a 0.25 w/w% of (+) 11 remained homogeneous, while at a concentration of 0.125 w/w%, it became a cloudy suspension rather than precipitation. The dilution is expected to shift the equilibrium: (R′)2R′′NH+ (11) ⇋ (R′)2R′′N: +H+ toward right, thereby increasing the fractional contribution of the water-insoluble neutral amine (R′)2R′′N: on the polymer chain. However, in salt-free water, 0.125 w/w % polymer remained soluble by shifting the equilibrium to the left, since the ammonium salt is a weaker acid in salt-free water than in 0.1 M NaCl [22]. As expected of the majority of electroneutral (±) polymers [25–28], zwitterionic 13 was found to be insoluble in salt-free water, but soluble in the presence of minimum critical salt concentrations (CSCs) of 4.73 M NaBr and 2.90 M NaI. The I− being more polarizable than Br− is more effective in neutralizing the ionic cross-links, thereby disrupting the intragroup, intra- and interchain attractive interactions [13]. Such large CSC values indicate the remarkable strength of the zwitterionic interactions. It is quite surprising that the polymer was found to be insoluble in the presence of any concentration of NaCl or HCl (0.1–12 M). We find no rationale behind the insolubility in the presence of HCl which is expected to push zwitterionic (±) 13 toward CPE (+) 12. It has been revealed during potentiometric titrations that addition of NaOH to an aqueous mixture of (±) 13 led to its solubility at a point where the backbone composition of repeat unit of (±) 13/(± −) 14 becomes less than 90:10. Increasing the anionic portion thus leads to solubility in water.
Infrared and NMR spectra
The carbonyl frequency of the ester group in CPE 11 is confirmed by the presence of the IR band at 1745 cm−1. The strong bands around 1306 and 1126 cm−1 were assigned to the respective asymmetric and symmetric vibrations of the SO2 unit in 11 as well as in 13. The symmetric and anti-symmetric stretching of COO− in the dipolar form 13 appeared at 1386 and 1628 cm−1 respectively, similar to those observed for simple amino acids [29], while the absorption for the C=O stretch of COOH appeared at 1728 cm−1.
The 1H and 13C NMR spectra of monomers 5 and 10, and polymers 11 and 13, displayed in Fig. 1 and 2, respectively, reveal the absence of any residual alkene in the polymers as a consequence of faster intramolecular cyclization than intermolecular propagation in the cyclopolymerization protocol. The chain termination process by abstraction of an allylic proton of the monomer [30], as well as coupling process [31], would also lead to the absence of residual alkene in the polymers.
Viscosity measurements
Viscosity data for CPE (+) 11 (entry 4, Table 1) are plotted in Fig. 3b–i. In salt-free water, the viscosity plot for 11 was not typical of polyelectrolytes (i.e., concave upward) (Fig. 3b–i); with decreasing concentration, the reduced viscosity decreases as a result of increasing presence of the neutral amine form in the mobile equilibrium: (R′)2R′′NH+ (11) ⇋ (R′)2R′′N: +H+ (vide supra). The viscosity plots move downward in 0.1 M NaCl as compared in salt-free water owing to the shielding of the cationic charges by Cl− ions [Fig. 3b-iii]. The straight-line plot changed direction downward at lower concentrations; the solution became a cloudy suspension at 0.125 w/w % as a result of the presence of excessive amount of the neutral amine. The reduced repulsive charges on the polymer chain therefore decrease the hydrodynamic volumes and hence decrease the viscosity values. The viscosity plot becomes normal in 0.1 M HCl (Fig. 3b-ii); added H+ forces CPE 11 to be in the cationic form. The equilibrium mentioned above is shifted toward left, and Cl− ions shield the positive nitrogens so as to give a normal linear plot. A comparison between Fig. 3b-ii and b-iii confirms that the higher viscosity in 0.1 M HCl than in 0.1 M NaCl is a result of the higher cationic charge density in the polymer chain in the former medium.
PZA (±) 13 is water insoluble; however, in the presence of 1 and 2 equivs. NaOH, it is converted into (± −) PZAN 14 and (=) PDE 15, respectively. The viscosity plots of 14 (Fig. 3c-i) and 15 (Fig. 3c-ii) are concave upward in salt-free water as expected of polyelectrolytes, while they became linear in 0.1 M NaCl (Fig. 3c-iii and iv). It is surprising to note that dianionic (=) 15 has lower viscosity values (Fig. 3c-ii) than zwitterionic/anionic (± −) 14 (Fig. 3c-i) in salt-free water, as confirmed by careful triplicate measurements under N2. As per expectation, (=) 15 should have higher viscosity values as a result of greater repulsion among its repeating units having higher negative charge density. The expected trend is observed in 0.1 M NaCl (Fig. 3c-iii versus iv), but not in salt-free water. One possible rationale behind these findings is that in salt-free water, the shorter distance between the positive nitrogens in the neighboring zwitterionic dipoles in (± −) 14 leads to greater repulsions than the repulsions experienced among more distant negative oxygens in the neighboring repeating units of dianions (=) 15. In 0.1 M NaCl, the Cl− ions effectively neutralize the positive nitrogens in (± −) 14, permitting greater negative charges in (=) 15 to dictate the viscosity values. To correlate two polymers (+) 11 (Table 1) and (±) 13 (Table 2), obtained via respective monomers 10 and 5, diester 11 was hydrolyzed to 13 (Scheme 2). The intrinsic viscosities of (± −) 14 [i.e., (±) 13 + 1 equiv NaOH] prepared from Table 1 (entry 4) and Table 2 (entry 1) were determined to be 0.354 and 0.585 dL g−1, respectively (cf. Fig. 3c-iv vs. c-v). Using Mark–Houwink equation ([η] = KMa) with an approximate ‘a’ value of 0.8, the sample from Table 2 (entry 1) is estimated to have an Mw of 105,000 g mol−1 as calculated from the known Mw of 56,000 g mol−1 for the sample of Table 1 (entry 4) and the [η] values of 0.354 and 0.585 dL g−1 as presented above. The diester monomer 10 was thus found to give polymers (Table 1) with lower molar masses as compared to its hydrolyzed counterpart 5 (Table 2), presumably as a result of higher steric crowding of the ester group (during cyclization) than its hydrolyzed counterpart.
Basicity constants
Basicity constants K 1, K 2 and K 3 describe the protonation of repeat units in (=) 15, (± −) 14 and (±) 13, respectively. Unlike small molecules, the K i of a repeat unit is ‘apparent’ [32]; it depends on the type of charge and its density in the neighboring units and as such varies with the degree of protonation (α). The K is of anionic centers is described by Eq. (3) (Scheme 2), where log K o i = pH at α = 0.5 and n i = 1 in the case of sharp basicity constants. Using Eq. (2), the pH vs. log [(1 − α)/α)] plots enabled us to determine the ‘n i ’ and log K o i as the slope and intercept, respectively, as described elsewhere [22]. In salt-free water, basicity constants log K 1 and log K 2 were determined to be 9.91 and 5.33, respectively, while the corresponding n 1 and n 2 were found to be 1.71 and 1.35 (Table 3). Log K 3 involving the equilibrium: 13 (\({\text{ZH}}_{2}^{ \pm }\)) + H+ ⇌ (\({\text{ZH}}_{3}^{ + }\) ) 12 could not be determined owing to the water insolubility of the component polymers. It was revealed during titration that a polymer backbone consisting of less than 90 % zwitterionic (\({\text{ZH}}_{2}^{ \pm }\)) 13 and more than 10 % zwitterionic-anionic (\({\text{ZH}}_{2}^{ \pm - }\)) 10 remained water soluble, while higher percentages of zwitterionic fraction impart insolubility.
Any value of n i (i.e., polyelectrolyte index) other than 1 leads to variation of K with α (Fig. 4a). The n i values greater than 1 lead to the decrease of K with α. A strong polyelectrolyte effect is reflected by a greater value of n associated with a greater variation of K with α. A gradual decrease of average negative charge density per repeating unit with increasing α reduces the electrostatic field that induces protonation. It is worth mentioning that log K of a base is the pK a of its conjugate acid.

a Plot for the apparent log K i versus degree of protonation (α) filled square (K 1, entry 1, Table 3) open square (K 2, entry 1, Table 3). b Precipitation behavior of a supersaturated solution of CaSO4 in the presence (20, 30 ppm) and absence of PZA 13 (+1 equiv. NaHCO3). c Phase diagram [filled square and open square represent data obtained by respective NMR and turbidity method] at 296 K of PZA 13 (treated with 2 equivs. NaOH)–UPVA–H2O (0.6 M NaCl) at 296 K. d Correlation of phase diagram of PZA 13 (2 equiv. NaOH)–UPVA–H2O (0.6 M NaCl)
Inhibition of CaSO4 scale formation by the synthesized polymer
In the reverse osmosis (RO) process, the inlet feed water stream is divided into product water and reject brine which may become supersaturated with salts that may lead to scaling, thus reducing the efficiency of the desalination membrane. The percent inhibition (PI) of scaling is calculated using Eq. (4):
where \([ {\text{Ca}}^{{ 2 { + }}} ]_{{{\text{inhibited(t}}_{ 0}) }}\), \([ {\text{Ca}}^{{ 2 { + }}} ]_{\text{inhibited(t)}}\) and \([ {\text{Ca}}^{{ 2 { + }}} ]_{\text{blank(t)}}\) are the Ca2+ concentrations at time zero and t in the inhibited and blank solution (without antiscalant), respectively.
For the current work, the scaling behavior of a supersaturated solution of CaSO4 containing 2598 ppm of Ca2+ and 6300 ppm of SO4 2− in the presence of 30 and 20 ppm of synthesized antiscalant 13 was investigated, and the results of percentage scale inhibition are given in Table 4. The effectiveness of an antiscalant depends on its ability to scavenge metal cations [7] and interfere with crystal formation at the time of nucleation [33]. In the absence of antiscalant, the precipitation of CaSO4 is indicated by a sudden drop in conductivity (Fig. 4b-iii: blank). To our satisfaction, the presence of 20 and 30 ppm 13 registered scale inhibitions of 98 and 99 %, respectively, for about 30 min as calculated using Eq. (4). In the presence of 30 ppm antiscalant, it registered a 97.5 % scale inhibition at a time of 1500 min. Usually, a residence time of ≈30 min is required for the brine in the osmosis chamber. At the inhibitor concentration of 20 ppm, an induction period of 70 min is followed by a sharp drop in conductivity, which indicates an accelerated growth of CaSO4 crystal (Fig. 4b-ii). However, at the time scale of over 3000 min, no induction was observed at the inhibitor concentration of 30 ppm.
In the presence of an antiscalant, crystals having irregular shapes and loose structures of smaller fragments are formed. The involvement of the antiscalant into the nucleation process during the induction period leads to the prevention of the normal growth of CaSO4·2H2O by poisoning the active growing sites in the crystal. This leads to the increase in the internal stress of the distorted crystals, thereby resulting in crystal fractures and prevention of deposition of microcrystals [33]. Later on, after all the antiscalant molecules are adsorbed into growing crystal, the crystal growth can resume at a rate comparable to that of the unpoisoned systems. So, the induction period depends on the concentration of the antiscalant; at a higher concentration (30 versus 20 ppm), it is able to prolong the induction period and may thus minimize the fouling of membranes by CaSO4·2H2O.
Phase diagrams using [PZA 13 + 2 equivs. NaOH]—UPVA 17–H2O (NaCl) systems
The turbidity method was used to construct the binodal, while 1H NMR technique was utilized for the tie lines in phase diagram of UPVE 17–PDE 15 (i.e., PZA 13 + 2 equiv NaOH)–0.6 M NaCl in Fig. 4c. The A total represents the composition of a total system, which splits into PZA-rich top (A top) and UPVA-rich bottom (A bottom) phases. The three compositions are connected by a tie line where volume ratio (or the mass ratio) of the top and bottom phases V top/V bottom is equated to the ratio of the tie-line length: (A total − A bot)/(A total − A top) [17]. Variation in the A total is helpful in the construction of ATPS with a suitable volume ratio.
A binodal curve demarcates a single- and two-phase region and thus provides information about the suitability of the polymers for industrial separation process. Its position closer to the axes makes the ATPS more economical, since it will require lower amount of polymers for phase separation to occur. For the current ATPS, the phase separation occurs at total polymer concentrations of ≈5 %, which is excellent from an industrial point of view. At higher polymer concentrations, ATPS systems become more expensive. For bioseparation, the high water content is expected to make this ATPS biocompatible and benign to biomaterials. One of the most gratifying aspects of the current ATPS is the solubility behavior of the component polymer PZA 13. Environmentally friendly use of ATPSs in separation and purification of biomolecules demands a way to their recycling [17], which is possible with the current polymer 13, since at lower pH values, it is practically insoluble in water. Though outnumbered by construction and use of non-ionic ATPSs, the application of ionic polymers in protein separation has also been documented [18]. PZA 13 containing aspartic acid residues with pH-triggerable functionalities (N and CO2 −) is anticipated to impart pH-responsive behaviors in selective separation and purification of biomolecules, like proteins.
The correlation of the phase diagrams
Equations (5) and (6), based on Florey–Huggins theory, were developed by Diamond and Hsu [34] to check the consistency of the tie lines.
and
where the subscripts 1 and 2 represent polymer 1 (UPVA) and polymer 2 (PZA 13 + NaOH), while w″ and w′ denote the polymer weight percent in the top and bottom phase, respectively. The magnitude of the slopes A 1 and A 2 reflects the effects of polymer molar masses and their interactions with water. The concentration ratio of the polymers in the top and bottom layer (C t/C b) represents the partition coefficient K 1 and K 2. The simple model thus describes the phase behavior reasonably well as shown by the good least square linear fits of the data in Fig. 4d. A linear regression of ln K versus \(w_{i}^{\prime \prime } - w_{i}^{\prime }\), with zero intercept value afforded the parameters A 1 and A 2. The root mean-square deviation (rmsd) was calculated using the experimental K exp and the calculated partition constants K cal by Eq. (7) [35]:
where the parameters Ai (A 1 = −0.371, A 2 = 1.51) and rmsd values [(rmsd)1 = 0.0257, (rmsd)2 = 2.87) of the correlation model for the partition coefficients using N number of tie lines (Fig. 4d) ascertain that Eqs. (5) and (6) are helpful in correlating the experimental data of the current work. The simple model thus requires only a single phase composition to calculate A 1 and A 2, thereby it helps to avoid extensive phase equilibrium determination.
Poly(N,N-diallylaspartic acid) 6 versus Poly(N,N-diallylaspartic acid-alt-SO2) 13: a comparison of the homo- and copolymers
Several properties of homopolymer poly(diallyl aspartate) (6) and copolymer poly(diallyl aspartate-alt-SO2) (13) are summarized in Table 6. The presence of electron-withdrawing SO2 unit has decreased the log K 1, i.e., log [basicity constant] of the amine nitrogen by 1.2 unit as compared to the homopolymer. Since log K 1 = pK +NH , the conjugate acid 14 of copolymer 15 is thus more acidic than its homopolymer counterpart. The presence of SO2 is also expected to increase the acidity of diester copolymer 11; its increased dissociation to the neutral amine form led to its insolubility in dilute aqueous mixture (vide supra). Butler’s cyclopolymerization protocol gave copolymers having much higher molar masses than the corresponding homopolymers (Table 6). Homopolymer 6 imparted much higher scale inhibition than copolymer 13; at a 20 ppm concentration, the inhibition of CaSO4 scale formation after the elapse of 1500 min was found to be 100 and 14 %, respectively. It is to be noted, in this respect, that PASP (3/4) (Scheme 1) with lower molar masses has been shown to impart better scale inhibition than the polymer with higher molar masses. PASP with molar masses in the range 1000–2000 g mol−1 imparted an inhibition efficiency of 80 % in a supersaturated 0.1 M Na2SO4 and CaCl2 solutions at 30 °C for 24 h [7]. Therefore, it is logical to conclude that the better scale inhibition by homopolymer 6 as compared to copolymer 13 must be a consequence of its lower molar mass. In future endeavor, attempts would be made to obtain homo- and copolymers with molar masses as low as possible to achieve better scale inhibition efficiencies. Polymers with smaller sizes can interfere more effectively with the nucleation and growth of scale.
There is a great difference in the solubility behaviors of the homo- and copolymers. The copolymer has higher CSC values for various salts i.e., higher amount of salts are required to promote its water solubility (Table 6). As a result of electron withdrawal by the SO2 unit, the positive charge on the nitrogens in copolymer 13 is more dispersed and hence less hydrated. The enhanced zwitterionic attraction in 13 in the absence of interference by hydration demands greater amount of salts for its disruption to promote solubility. It is interesting to note that the copolymer is practically insoluble in the presence of any amount of added HCl or NaCl, thereby making it a suitable polymer component in a pH-controlled recyclable ATPS.
Conclusions
Monomers 5 and 10 containing residue of aspartic acid have been cyclopolymerized with SO2 to obtain alternate copolymers PZA 13 (i.e., 5-alt-SO2) and CPE 11 (i.e., 10-alt-SO2) in very good yields. PZA 13 is practically insoluble in water or aqueous HCl or NaCl, while soluble only in the presence of high concentrations of NaBr and NaI. The addition of NaOH transforms PZA (±) 13 to water-soluble PZAN (± −) 14 and PDE (=) 15, which has been used as a component polymer in the construction of a recyclable ATPS. The interesting solubility behavior envisages its use at a higher pH value and recycling by precipitating at a lower pH. The basicity constant K for the ligand centers in the following equilibria (=) 15 + H+ ⇋ (± −) 14 and (± −) 14 + H+ ⇋ (±) 13 have been determined; the data would be of great value for proper utilization of the polymer as a scavenger of toxic metal ions and inhibitor of metal corrosion.
The potential of PZA 13 as an antiscalant has been demonstrated; it imparted excellent CaSO4-scale inhibition. The interesting pH-dependent solubility behavior for the current polymers could well be exploited in forming blend films containing zwitterion structure units for potential application in membrane separation [36]. Work is currently underway in our laboratory to utilize current monomers to synthesize aspartic acid-based cross-linked resin as chelating adsorbents for the removal of toxic materials.
Reference
Krebs HA, Weitzman PDJ (1987) Krebs’ citric acid cycle: half a century and still turning. Biochemical Society, London. ISBN 0-904498-22-0
Roweton S, Huang SJ, Swift G (1997) Poly (aspartic acid)—synthesis, biodegradation, and current applications. J Environ Polym degrad 5:175–181
Rusenko KW, Donachy JE, Wheeler AP (1991) Purification and characterization of a shell matrix phosphoprotein from the american oyster. In: Sikes CS, Wheeler AP (eds) Surface reactive peptides and polymers. ACS Symposium Series 444. ACS, pp 107–124
Joentgen W, Müller N, Mitschker A, Schmidt H (2004) Polyaspartic acids. In: Fahnestock S, Steinbüchel A (eds) Polyamides and complex proteinaceous materials I. Biopolymers 7. Wiley-VCH. pp 175–179. ISBN 9783527302222
Low KC, Wheeler AP, Koskan LP (1996) 6: Commercial poly (aspartic acid) and its uses. In: Glass JE (ed) Hydrophilic polymers, advances in chemistry 248, ACS pp 99–111
Gross RA, Kalra B (2002) Biodegradable polymers for the environment. Science 297(5582):803–807
Hasson D, Shemer H, Sher A (2011) State of the art of friendly “green” scale control inhibitors: a review article. Ind Eng Chem Res 50:7601–7607
Zahuriaan-Mehr MJ, Pourjavadi A, Salimi H, Kurdtabar M (2009) Protein-and homo poly (amino acid)-based hydrogels with super-swelling properties. Polym Adv Technol 20:655–671
Thombre SM, Sarwade BD (2005) Synthesis and biodegradability of polyaspartic acid: a critical review. J Macromol Sci Part A 42:1299–1315
Liu M, Wang L, Su H, Cao H, Tan T (2013) pH-sensitive IPN hydrogel based on poly (aspartic acid) and poly (vinyl alcohol) for controlled release. Polym Bull 70:2815–2827
Butler GB (1992) Cyclopolymerization and cyclocopolymerization. Marcel Dekker, New York
Ali SA, Zaidi SMJ, El-Sharif AMZ, Al-Taq Ali A (2012) Cyclopolymers from N, N-Diallyl-N-propargyl-(12-N′-formylamino)-1-dodecylammonium chloride and Their use as Inhibitors for mild steel corrosion. Polym Bull 69:491–507
Kudaibergenov S, Jaeger W, Laschewsky A (2006) Polymeric betaines: synthesis, characterization, and application. Adv Polym Sci 201:157–224
Singh PK, Singh VK, Singh M (2007) Zwitterionic polyelectrolytes: a review. E-Polymers 030:1–34
Jamiu ZA, Al-Muallem HA, Ali SA (2015) Aspartic acid in a new role: synthesis and application of a pH-responsive cyclopolymer containing residues of the amino acid. React Func Polym 93:120–129
Al-Muallem HA, Wazeer MIM, Ali SA (2002) Synthesis and solution properties of a new pH-responsive polymer containing amino acid residues. Polymer 43:4285–4295
Albertsson P (1986) Partition of cell particles and macromolecules, 3rd edn. Wiley, Newyork
Waziri M, Abu-Sharkh BF, Ali SA (2004) Protein partitioning in aqueous two-phase systems composed of a pH-responsive copolymer and poly (ethylene glycol). Biotechnol Prog 20:526–532
Ali SA, Al-Hamouz OCS, Hassan NM (2013) Novel cross-linked polymers having pH-responsive amino acid residues for the removal of Cu2+ from aqueous solution at low concentrations. J Hazard Mater 248–249:47–58
Ali SA, Al-Muallem HA, Mazumder MAJ (2013) Coexistence curves of aqueous two-phase systems of some pH-responsive homo- and copolymers of 3-(diallylammonio)propane-1-sulfonate and urethanized poly (ethenol) or poly (oxyethylene). J Chem Eng Data 58:2574–2585
Andrews BA, Asenjo JA (2010) Theoretical and experimental evaluation of hydrophobicity of proteins to predict their partitioning behavior in aqueous two phase systems: a review. Sep Sci Technol 45:2165–2170
Ali SA, Abu-Thabit NY, Al-Muallem HA (2010) Synthesis and solution properties of a pH-responsive cyclopolymer of zwitterionic ethyl 3-(N, N-diallylammonio) propanephosphonate. J Polym Sci Part A Polym Chem 48:5693–5703
Haladu SA, Ali SA (2013) A pH-responsive cyclopolymer having phospho- and sulfopropyl pendents in the same repeating unit: synthesis, characterization, and its application as an antiscalant. J Polym Sci Part A Polym Chem 51:5130–5142
Rullens F, Devillers M, Laschewsky A (2004) New regular, amphiphilic poly (ampholyte)s: synthesis and characterization. Macromol Chem Phys 205:1155–1166
Wielema TA, Engberts JBFN (1987) Zwitterionic polymers—I. Synthesis of a novel series of poly (vinylsulphobetaines). Effect of structure of polymer on solubility in water. Eur Polym J 23:947–950
Salamone JC, Volksen W, Olson AP, Israel SC (1978) Aqueous solution properties of a poly (vinyl imidazolium sulphobetaine). Polymer 19:1157–1162
Yoshizawa M, Ohno H (1999) Molecular brush having molten salt domain for fast ion conduction. Chem Lett 889–890
Laschewsky A (2014) Structures and synthesis of zwitterionic polymers. Polymers 6:1544–1601
Pearson JF, Slifkin MA (1972) The infrared spectra of amino acids and dipeptides. Spectrochim Acta A 28A:2403–2413
Butler GB, Angelo RJ (1957) Preparation and polymerization of unsaturated quaternary ammonium compounds. VIII. A proposed alternating intramolecular–intermolecular chain propagation1. J Am Chem Soc 79:3128–3131
Pike RM, Cohen RA (1960) Organophosphorus polymers. I. Peroxide-initiated polymerization of diethyl and diisopropyl vinylphosphonate. J Polym Sci 44:531–538
Barbucci R, Casolaro M, Danzo N, Barone V, Ferruti P, Angeloni A (1983) Effect of different shielding groups on the polyelectrolyte behavior of polyamines. Macromolecules 16:456–462
Davey RJ (1982) The role of additives in precipitation processes. In: Jancic SJ, de Jong EJ (eds) Industrial crystallization 81. North-Holland, Amstardam, pp 123–135
Diamond AD, Hsu JT (1992) Correlation of polymer partitioning in aqueous two-phase systems. AIChE Symp Ser 290:105–111
Shahriari S, Doozandeh SG, Pazuki G (2012) Partitioning of cephalexin in aqueous two-phase systems containing poly (ethylene glycol) and sodium citrate salt at different temperatures. J Chem Eng Data 57:256–262
Zhang Y, Zhou Y, Liu C (2014) Structure and properties of novel blend films from alginate and quaternized poly(4-vinyl-N-carboxymethylpyridine) containing zwitterion structure units. Polym Bull 71:2014
Acknowledgments
This project was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz City for Science and Technology, through the Science & Technology Unit at King Fahd University of Petroleum & Minerals (KFUPM), the Kingdom of Saudi Arabia, award number (11-ADV2132-04). The authors gratefully acknowledge the facilities provided by KFUPM.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ali, S.A., Jamiu, Z.A. Poly(N,N-diallylaspartic acid-alt-sulfur dioxide): its synthesis and application. Polym. Bull. 73, 2179–2198 (2016). https://doi.org/10.1007/s00289-016-1602-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00289-016-1602-x