
Abstract
Background
Infants born via cesarean section (CS) are at an increased risk of immune-related diseases later in life, potentially due to altered gut microbiota. Recent research has focused on the administration of probiotics in the prevention of gut microbiota dysbiosis in neonates delivered by CS. This study was performed to investigate the effects of probiotic supplementation on the gut microbiota of CS-delivered infants.
Methods
Thirty full-term neonates delivered by CS were randomized into the intervention (supplemented orally with a probiotic containing Bifidobacterium longum, Lactobacillus acidophilus, and Enterococcus faecalis for 2 weeks) and control groups. Stool samples were collected at birth and 2 weeks and 42 days after birth. The composition of the gut microbiota was analyzed using 16S rRNA sequencing technology.
Results
The applied bacterial strains were abundant in the CS-delivered infants supplemented with probiotics. Probiotics increased the abundance of some beneficial bacteria, such as Bacteroides, Acinetobacter, Veillonella, and Faecalibacterium. Low colonization of Klebsiella, a potentially pathogenic bacterium, was observed in the intervention group.
Conclusions
Our results showed that probiotics supplemented immediately after CS enriched the gut microbiota composition and altered the pattern of early gut colonization.
Trial Registration: registration number NCT05086458.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colonization of the infant-gut microbiota begins at birth and is considered to be a dynamic and finely regulated process during the first years of life [1]. Infants may receive specific microbial signals in a critical developmental time window. It is well known that the gut microbiota critically influences the development and function of the immune [2], metabolic [3], gastrointestinal [4], and nervous systems [5].
Infant microbiota is very unstable and can be influenced by many factors, such as the delivery mode. The natural colonization and development of infant-gut microbiota is stunted when the infant is born by cesarean section (CS) [6]. A longitudinal study of 150 countries showed an increasing trend in CS rates from 6.7% in 1990 to 19.1% in 2014 [7]. In addition to the maternal and neonatal risks associated with CS, it also leads to dysbiosis of the infant gut microbiota, possibly challenging long-term health, including allergies [8], immunological diseases [9], and metabolic disorders [10]. The increasing rate of CS has raised a significant public health concern due to the disruption of early life microbiota; however, to date, specific treatment modalities are still lacking. It is increasingly apparent that preventing or decreasing disruptive effects on the gut microbiota is important for the healthy development of infants.
As shown in our previous study [11], delayed intestinal colonization of Bifidobacterium was observed in CS-born infants, which is consistent with previous clinical studies [12, 13]. Moreover, the colonization of Lactobacillus is significantly damaged in CS-born infants compared with those delivered vaginally [14]. Al-Balawi et al. [15] showed that Enterococcus faecalis was the most dominant, representing more than 60% of the total lactic acid bacteria in the initial colonization of healthy newborns in the first week of their life. Early colonizers play an important role in immune system development and provide colonization resistance by preventing the overgrowth of opportunistic pathogens [16]. Therefore, attempts to regulate the gut microbiota in newborns delivered by CS with probiotics representing important bacterial species in early life after birth have been made. The aim of the present study was to investigate the effect of postnatal supplementation with a multispecies probiotic (Bifidobacterium longum, Lactobacillus acidophilus, and Enterococcus faecalis) on the global gut microbiota composition in CS-delivered infants.
Materials and Methods
Patients and Study Design
This was an exploratory, randomized, open-label, parallel-controlled study conducted at the Department of Pediatrics, Shanghai Tenth People’s Hospital, Tongji University School of Medicine from August 2021 to December 2021. Informed consent was obtained from all guardians of the enrolled neonates before randomization. This protocol was approved by the Ethics Committee of Shanghai Tenth People’s Hospital (approval no. SHSY-IEC-4.1/21–188/01) and adhered to the tenets of the Declaration of Helsinki. This study was registered at Clinicaltrials.gov (NCT05086458).
Only neonates born via elective CS and those whose parents had decided to exclusively feed them with breast milk half an hour after birth were assessed for eligibility to participate in the study. A total of 35 consecutive neonates were enrolled in the study according to the following criteria: (1) primipara mothers aged between 25 and 35 years without pregnancy complications; (2) all mothers did not receive other antibiotics apart from a single intravenousdose of 3.0 g cefuroxime, given before CS; (3)2500 g ≤ birth weight < 4000 g, 37 weeks ≤ gestational weeks < 42 weeks, without a history of asphyxia at birth; (4) infant only receive breastfeeding within 42 days after birth;and (5) being 24 h old or younger at the time of enrollment. The exclusion criteria were: (1) mother took probiotic supplements during delivery; (2) infant had congenital metabolic or hereditary disease; (3) infant had been treated with antibiotics and participated in another study; (4) infant had any significant prenatal or postnatal disease; and (5) a lack of informed consent by the parents, or their resignation from the study. A restricted block randomization sequence was created with 1:1 allocation using a fixed block size of four. The block size was unknown to both the investigators and the participants. A data manager who was not associated with the clinical portion of this study prepared the randomization sequence using computer-generated random numbers. Eligible neonates were randomly assigned to receive either probiotic compounds (probiotic group:within 24–48 h after birth, 0.5 g per treatment, three times per day, for 2 weeks) or no other intervention (control group). The probiotic supplement (BIFICO, Shanghai Sinepharm, China) comprised > 1.0 × 107 CFU of Bifidobacterium longum, Lactobacillus acidophilus, and Enterococcus faecalis per gram.
Fecal samples were collected at three points in time: newborn (T0) and 2 weeks (T1) and 42 days (T2) after birth. Each fecal sample was collected in a sterile tube and then stored at − 80 °C prior to microbial analysis. Clinical information to be used in the analysis, including gestational age, sex, and birth weight, were retrieved from the digital medical records system.
DNA Extraction, Amplification, and Bioinformatics Analysis
The procedures used in this study are described in a previous study [17]. In brief, DNA was extracted from fecal samples using the E.Z.N.A.® Soil DNA kit (Omega Bio-tek, Norcross, GA, USA) according to the manufacturer’s instructions and was quantified using a NanoDrop 2000 UV–vis spectrophotometer (Thermo Fisher Scientific, Wilmington, MA, USA). The hypervariable V3-4 regions of the 16S rRNA gene in the gut microbiota were amplified by polymerase chain reaction using specific primers and sequenced. The primers used for PCR amplification in the V3-V4 region were 338f (5′-ACTCCTACGGGGGGCAGG-3′) and 806r (5′-GACTACHVGGGTWTCTAAT-3′), with an amplification length of approximately 460 bp. The amplified 16S rRNA amplicons were then purified using a DNA gel extraction kit (Axygen Biosciences, Union City, CA, USA) and sequenced using the Illumina MiSeq platform (Illumina, San Diego, CA, USA). The raw FASTQ files were demultiplexed, quality-filtered using Trimmomatic, and merged with FLASH. Operational taxonomic units (OTUs) were clustered at a 3% divergence (97% similarity). Chimeric sequences were identified and removed using UCHIME (version 4.2.40; http://drive5.com/usearch/manual/uchime_algo.html). A taxonomic analysis of the representative sequences of each OTU was performed. The RDP Classifier algorithm (http://rdp.cme.msu.edu/) was used to analyze the taxonomy of each 16S rRNA gene sequence. Comparisons of the richness and diversity of the microbial communities were performed after OTU identification. The taxa that were differentially enriched in each group were identified using linear discriminant analysis coupled with effect size (LEfSe). Differences in the microbial structure were evaluated using principal coordinate analysis. The number of permutations used to compare microbial differences was set to 999. The Cytoscape platform (version 3.4.0; http://www.cytoscape.org/) was used for co-abundance analysis.
Statistical Analyses
All statistical analyses were performed using PASW SPSS 22.0 (IBM, Armonk, NY, USA) and GraphPad Prism 7.00 (GraphPad Software, San Diego, CA, USA). Continuous variables were expressed as means ± standard deviations. We used Student’s t and Pearson’s chi-square tests to analyze and compare the continuous and categorical variables, respectively. Mann–Whitney U rank tests were used to compare the differences between two groups. Differences were considered significant at P < 0.05.
Availability of Data and Materials
The datasets generated and/or analysed during the current study are available in the National Center for Biotechnology Information repository, Sequence Read Archive(SRA)database (https://www.ncbi.nlm.nih.gov/sra/) and the accession number is SRP134214.
Results
Baseline Characteristics of Enrolled Neonates
A total of 35 eligible newborns were included in the study. Sixteen neonates were enrolled in the intervention group, whereas the others were enrolled in the control group. Two newborns were excluded from the intervention group after 42 days because the stool sample was not provided; thus, the sample size for this group was 14. For the same reason, the final number of newborns in the control group was 16, as shown in Table 1.
Comparisons between the groups showed that there were no significant differences with respect to gestational age, sex, birth weight, feeding mode, or maternal antibiotic treatment before or during labor. None of the newborns in the intervention group experienced side effects caused by probiotics, such as abdominal distension, diarrhea, vomiting, and sepsis.
Community Richness and Diversity
To characterize the gut microbiota in CS-delivered infants supplemented with probiotics, we compared the alpha diversity between the probiotic and control groups. At birth, there were no significant differences in bacterial richness and diversity between the two groups, as shown in Supplementary Fig. 1 (Online Resource 1). We found significantly higher bacterial richness at week 2 (Fig. 1a and b) and day 42 (Fig. 1e and f) in the probiotic group and no significant difference in bacterial diversity between the two groups (Fig. 1c, d, g and h).

Comparison of the alpha diversity at T1 and T2 between the control and probiotic group. Ace index at T1 (a), *P < 0.05; Chao index at T1 (b), **P < 0.01; Shannon index at T1 (c), P = 0.12 (>0.05); Simpson index at T1 (d), P = 0.33(>0.05). Ace index at T2 (e), ****P < 0.0001; Chao index at T2 (f), ****P < 0.0001; Shannon index at T1 (g), P = 0.17(>0.05); Simpson index at T1 (h), P = 0.41 (> 0.05)
Significant Difference in Microbiota Between the Probiotic and Control Groups
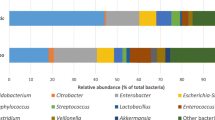
To establish an overall effect of the probiotics, we tested whether the supplement ameliorated some of the CS-induced changes in microbiota composition. We further explored the relative taxon abundance in the microbiota of the probiotic and control groups. The total distribution of bacterial taxa showed a significant variation between the two groups at the class level (Fig. 2a and b), as characterized by a significant decrease in the relative abundance of Gammaproteobacteria in the probiotic group compared with the control group (Fig. 3c and d). Clostridia increased significantly in the probiotic group at week 2 (Fig. 2a and c), whereas it did not differ significantly on day 42 (Figs. 2b and 3d).

Dominant phyla at T1 (a) and T2 (b); dominant genera at T1 (c) and T2 (d)

LEfSe analysis (a T1; b T2) and PCoA (c T1; d T2) between the control and probiotic group. The LDA score indicates the effect size and ranking of each differentially abundant taxon. Plot of PCoA scores based on the relative abundance of OTUs (97% similarity level). Each symbol represents a sample. R2 and P were calculated by the Adonis method. LDA linear discriminant analysis, LEfSe linear discriminant effect size, OTUs operational taxonomic units, PCoA principal coordinate analysis
We also compared differences at the genus level (Fig. 2c and d). Notably, a significant increase in the relative abundance of Enterococcus was observed in the probiotic group at week 2 (Fig. 3c), whereas Klebsiella decreased significantly at day 42 (Fig. 3d).
Unweighted PCoA showed that the microbiota of the probiotic group was distinct from that of the control group (Fig. 3a and b). LEfSe analysis was used to identify specific bacteria that were enriched in different groups. At week 2, Veillonella, Enterococcus, Clostridium, Lactobacillus, Bifidobacterium, and Acinetobacter (at the genus level) were dominant in the probiotic group, as indicated by the linear discriminant analysis (LDA) (LDA score > 3, Fig. 3c). At day 42, Clostridium, Lactobacillus, Actinomyces, Enterococcus, Bacteroides, Faecalibacterium, and Ralstonia (at the genus level) were enriched in the probiotic group (LDA score > 3, Fig. 3d). LEfSe analysis was used to identify specific bacteria that were enriched in same group. In the control group,Staphylococcus, Klebsiella, and Veillonella(at the genus level) were dominant in the second week, while Bifidobacterium and Haemophilus (at the genus level) were dominant at day 42 (LDA score > 3, Supplementary Fig. 2a). In the probiotic group, Staphylococcus and Veillonella(at the genus level) were predominant in the second week, while Escherichia,Lactobacillus,Bifidobacterium,Actinomyces,Propionibacterium,Bacteroides, Faecalibacterium, Rhodococcus,and Roseburia (at the genus level) were dominant at day 42 (LDA score > 3, Supplementary Fig. 2b).
Discussion
Many recent studies have confirmed that the establishment of the gut microbiota can be affected and disturbed by many environmental factors, including the mode of delivery [11,12,13, 18,19,20]. This early microbial community has been considered particularly sensitive to potential modulation by probiotic interventions, especially in infants delivered by CS [21]. However, the effect of probiotic administration on CS microbiota colonization is poorly understood. There is currently no clear consensus recommending the use of probiotics in CS-delivered neonates. To our knowledge, this is the first exploratory report on gut microbiota changes in population and diversity to examine the effect of probiotic supplementation in Chinese infants delivered by CS. We found that the gut microbiota of Chinese CS-born infants supplemented with probiotics showed significant changes, including increased bacterial richness and different microbiota structures.
In our study, we confirmed previous data showing perturbations of the early period of intestinal colonization in infants delivered by CS. The low abundance of Bifidobacterium, Lactobacillus, Acinetobacter, and Bacteroides, which are ubiquitous in the early microbiota of CS-born infants, were partly corrected by probiotic supplementation at different study ages. At week 2, in addition to the increase in Bifidobacterium, Lactobacillus, and Acinetobacter, there was a significant increase in Enterococcus. Forty-two days after birth, Lactobacillus was dominant in the probiotic group, followed by Actinomyces, Enterococcus, Bacteroides, Faecalibacterium, and Ralstonia. But the Bifidobacterium abundance was low. However,from day 1 to day 42, we found that after 42 days the Bifidobacterium abundance was high in both the control and intervention groups. Although different probiotics have been used, a higher abundance of Bifidobacterium or Lactobacillus has been consistently detected in similar studies [22, 23]. A recent study confirmed that prebiotic supplementation enhanced and sustained the successful colonization of lactobacilli [24]. Another study reported that gut colonization with Bifidobacterium and Lactobacillus was achieved in a few days and lasted for 1 month, with immediate supplementation of a probiotic containing Bifidobacterium breve PB04 and Lactobacillus rhamnosus KL53A for a few days in CS-born neonates. However, this study used standard quantitative cultures for Bifidobacterium and Lactobacillus, which only provided a partial understanding of the gut microbiota changes. In our study, gut colonization with Lactobacillus and Enterococcus was observed 2 weeks after starting supplementation and persisted consistently at 42 days after birth. Nevertheless, the higher abundance of Bifidobacterium was not sustained up to 42 days after birth. Three explanations for these discrepancies can be offered: (1) Compared with lactic acid bacteria, the colonization capacity of Bifidobacterium is not strong enough; (2) feeding with probiotics was too short or in a small dose; (3) the abundance of Bifidobacterium increased with time. It should be noted that there is currently no good method for restoring the Bacteroides population in CS-born infants [25]. Swabbing infants born by CS with the mother’s vaginal secretions have been shown to fail at Bacteroides restoration [26].However, a recent study observed maternal FMT does restore Bacteroides [27]. In the present study, gut colonization with Bacteroides was observed in the probiotic intervention groups. Our findings suggest that early intervention with probiotics can contribute to the fast recovery of the early microbiota dysbiosis induced by CS.
The most remarkable finding of our study is that the abundance of Veillonella and Faecalibacterium increased at the end of the intervention (2 weeks) and at 42 days after birth, respectively. One study has shown that the relative abundance of Veillonella and Faecalibacterium was significantly decreased in children at risk of asthma during early life [28]. Delivery by CS has been confirmed to be associated with childhood asthma [29]. The microbial hypothesis considers gut microbiota as the link between environmental changes and the immune system, and several recent studies have confirmed gut microbiota as a potential therapeutic target for preventing asthma and atopic diseases [30,31,32,33,34]. Although the purpose of the present study was not to measure any clinical endpoints as the primary outcome, our results suggest that early probiotic intervention could be expected to reduce the risk of asthma later in life, which needs to be confirmed by further studies.
In addition, bacteria considered to be potential pathogens were present in both the probiotic and control groups. Members of the Enterobacteriaceae family, such as Klebsiella, have been described as predominant in the gut of infants delivered by CS [12, 13], which was also confirmed in our previous study [11]. In the present study, the apparent effect of CS was the relative elevation in Klebsiella, gut colonization with Klebsiella was observed in the control groups. Nevertheless, the abundance of Klebsiella was low in the intervention groups, which was ameliorated by probiotic supplementation. Colonization of Klebsiella is not an unfamiliar phenomenon in early life and is usually controlled by commensal bacteria in the gut ecosystem. Although the exact mechanism of decreased Klebsiella infection remains to be clarified, it is speculated that the inhibition of pathogen colonization is determined by direct or indirect mechanisms of colonization resistance [35]. Several previous studies have demonstrated the antimicrobial activity of Bifidobacterium strains against groups of coliforms, including the genus Klebsiella [36, 37]. Our results suggested that the decrease in Klebsiella was probably due to the presence of the probiotic mixture itself in the fecal samples concomitantly with the stimulation of commensal bacteria, such as Bifidobacterium. Low et al. [38] observed that a high ratio of Klebsiella/Bifidobacterium in early life correlates with the later development of allergies in childhood. Although the implications of Klebsiella colonization for allergic diseases are limited, bacteria belonging to Klebsiella are known to be involved in the induction of pro-inflammatory responses in the host. For example, K. pneumoniae was shown to be highly associated with the colitogenic phenotype in a mouse model of inflammatory bowel disease [39]. A similar link between Klebsiella and intestinal inflammation has also been reported in infants with colic [40, 41]. Accordingly, early probiotic administration may improve certain immune phenotypes that are particularly relevant for CS-born infants.
It should be noted, however, that in both the 2-week and 42-day CS-infant samples, the probiotic supplementation failed to reduce the abundance of the genus Clostridium, including potential pathogenic species, such as Clostridium perfringens and Clostridium butyricum. However, intestinal colonization with Clostridium is very frequent and usually asymptomatic during the neonatal period [42, 43], Some Clostridium taxa might have beneficial immunoregulatory properties [44]. In our study, no infection symptoms were observed in the enrolled infants, and Clostridium abundance was higher in the probiotic group. A well-designed clinical study is warranted to confirm this observation.
There are some limitations to our study. First, the sample was not of sufficient size to comprehensively clarify the effects of probiotics on the gut microbiota. Second, in order to describe the effect of probiotic intervention on the gut microbiota, this study was limited to observing changes in the composition of the microbiota and could not exclude modifications of gene expression in the gut microbiota. Third, our follow-up did not cover a sufficient period for the long-term clinical or microbiological effects of probiotic intervention to be comprehensively observed. These limitations are expected to be overcome by more clinical and well-designed, longer follow-up studies.
Conclusion
Supplementation with a probiotic mixture containing Bifidobacterium longum, Lactobacillus acidophilus, and Enterococcus faecalis promoted the recovery of the microbiota dysbiosis of beneficial versus pathogenic bacteria induced by CS. These findings suggest that, in the absence of a normal pattern of colonization, early intervention with probiotics can generate substantial beneficial regulation of the gut microbiota. However, further studies are required to confirm these results.
References
Derrien M, Alvarez AS, de Vos WM (2019) The gut microbiota in the first decade of life. Trends Microbiol 27(12):997–1010
Gensollen T, Iyer SS, Kasper DL, Blumberg RS (2016) How colonization by microbiota in early life shapes the immune system. Science 352(6285):539–544
Blanton LV, Charbonneau MR, Salih T, Barratt MJ, Venkatesh S, Ilkaveya O, Subramanian S, Manary MJ, Trehan I, Jorgensen JM et al (2016) Gut bacteria that prevent growth impairments transmitted by microbiota from malnourished children. Science 351(6275):aad3311
Hooper LV, Gordon JI (2001) Commensal host-bacterial relationships in the gut. Science 292(5519):1115–1118
Yousefi B, Babaeizad A, Banihashemian SZ, Feyzabadi ZK, Dadashpour M, Pahlevan D, Ghaffari H, Eslami M (2022) Gastrointestinal tract, microbiota and multiple sclerosis (MS) and the link between gut microbiota and CNS. Curr Microbiol 80(1):38
Shao Y, Forster SC, Tsaliki E, Vervier K, Strang A, Simpson N, Kumar N, Stares MD, Rodger A, Brocklehurst P et al (2019) Stunted microbiota and opportunistic pathogen-34colonization in caesarean-section birth. Nature 574(7776):117–121
Betrán AP, Ye J, Moller AB, Zhang J, Gülmezoglu AM, Torloni MR (2016) The increasing trend in caesarean section rates: global, regional and national estimates: 1990–2014. PLoS ONE 11(2):e0148343
Papathoma E, Triga M, Fouzas S, Dimitriou G (2016) Cesarean section delivery and development of food allergy and atopic dermatitis in early childhood. Pediatr Allergy Immunol 27(4):419–424
Francino MP (2018) Birth mode-related differences in gut microbiota colonization and immune system development. Ann Nutr Metab 73(Suppl 3):12–16
Li N, Liang S, Chen Q, Zhao L, Li B, Huo G (2021) Distinct gut microbiota and metabolite profiles induced by delivery mode in healthy Chinese infants. J Proteomics 232:104071
Yang R, Gao R, Cui S, Zhong H, Zhang X, Chen Y, Wang J, Qin H (2019) Dynamic signatures of gut microbiota and influences of delivery and feeding modes during the first 6 months of life. Physiol Genomics 51(8):368–378
Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H et al (2015) Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17(5):690–703
Dogra S, Sakwinska O, Soh SE, Ngom-Bru C, Brück WM, Berger B, Brüssow H, Lee YS, Yap F, Chong YS et al (2015) Dynamics of infant gut microbiota are influenced by delivery mode and gestational duration and are associated with subsequent adiposity. MBio 6(1):e02419-e2514
Nagpal R, Tsuji H, Takahashi T, Kawashima K, Nagata S, Nomoto K, Yamashiro Y (2016) Sensitive quantitative analysis of the meconium bacterial microbiota in healthy term infants born vaginally or by cesarean section. Front Microbiol 7:1997
Al-Balawi M, Morsy FM (2020) Enterococcus faecalis is a better competitor than other lactic acid bacteria in the initial colonization of colon of healthy newborn babies at first week of their life. Front Microbiol 11:2017
Lundell AC, Bjornsson V, Ljung A, Ceder M, Johansen S, Lindhagen G, Törnhage CJ, Adlerberth I, Wold AE, Rudin A (2012) Infant B cell memory differentiation and early gut bacterial colonization. J Immunol 188(9):4315–4322
Gao R, Kong C, Huang L, Li H, Qu X, Liu Z, Lan P, Wang J, Qin H (2017) Mucosa-associated microbiota signature in colorectal cancer. Eur J Clin Microbiol Infect Dis 36:2073–2083
Xie J, Tang C, Hong S, Xin Y, Zhang J, Lin Y, Mao L, Xiao Y, Wu Q, Zhang X, Shen H (2023) Maternal vaginal fluids play a major role in the colonization of the neonatal intestinal microbiota. Front Cell Infect Microbiol 13:1065884
Reyman M, van Houten MA, van Baarle D, Bosch AATM, Man WH, Chu MLJN, Arp K, Watson RL, Sanders EAM, Fuentes S et al (2019) Impact of delivery mode-associated gut microbiota dynamics on health in the first year of life. Nat Commun 10(1):4997
Lu S, Huang Q, Wei B, Chen Y (2020) Effects of β-lactam antibiotics on gut microbiota colonization and metabolites in late preterm infants. Curr Microbiol 77(12):3888–3896
Walker WA (2013) Initial intestinal colonization in the human infant and immune homeostasis. Ann Nutr Metab 63(suppl 2):8–15
Rodenas CLG, Lepage M, Ngom-Bru C, Fotiou A, Papagaroufalis K, Berger B (2016) Effect of formula containing Lactobacillus reuteri DSM 17938 on fecal microbiota of infants born by cesarean-section. J Pediatr Gastroenterol Nutr 63(6):681–687
Bazanella M, Maier TV, Clavel T, Lagkouvardos I, Lucio M, Maldonado-Gòmez MX, Autran C, Walter J, Bode L, Schmitt-Kopplin P et al (2017) Randomized controlled trial on the impact of early-life intervention with bifidobacteria on the healthy infant fecal microbiota and metabolome. Am J Clin Nutr 106(5):1274–1286
Kongnum K, Taweerodjanakarn S, Hongpattarakere T (2020) Impacts of prebiotic-supplemented diets and breastmilk on population and diversity of lactobacilli established in thai healthy infants. Curr Microbiol 77(7):1191–1202
Korpela K, Salonen A, Vepsäläinen O, Suomalainen M, Kolmeder C, Varjosalo M, Miettinen S, Kukkonen K, Savilahti E, Kuitunen M et al (2018) Probiotic supplementation restores normal microbiota composition and function in antibiotic-treated and in caesarean-born infants. Microbiome 6(1):182
Dominguez-Bello MG, Jesus-Laboy KMD, Shen N, Cox LM, Amir A, Gonzalez A, Bokulich NA, Song SJ, Hoashi M, Rivera-Vinas JI et al (2016) Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat Med 22(3):250–253
Korpela K, Helve O, Kolho KL, Saisto T, Skogberg K, Dikareva E, Stefanovic V, Salonen A, Andersson S, de Vos WM (2020) Maternal fecal microbiota transplantation in cesarean-born infants rapidly restores normal gut microbial development: a proof-of-concept study. Cell 183(2):324-334.e5
Arrieta MC, Stiemsma LT, Dimitriu PA, Thorson L, Russell S, Yurist-Doutsch S, Kuzeljevic B, Gold MJ, Britton HM, Lefebvre DL et al (2015) Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci Transl Med 7(307):307ra152
Stokholm J, Thorsen J, Blaser MJ, Rasmussen MA, Hjelmsø M, Shah S, Christensen ED, Chawes BL, Bønnelykke K, Brix S, Mortensen MS, Brejnrod A, Vestergaard G, Trivedi U, Sørensen SJ, Bisgaard H (2020) Delivery mode and gut microbial changes correlate with an increased risk of childhood asthma. Sci Transl Med 12(569):eaax9929
Niu X, Yin X, Wu X, Zhang Q, Jiang Y, He J, Zhao Y, Zhang C, Ren Y, Lai M et al (2023) Heat-killed bifidobacterium longum BBMN68 in pasteurized yogurt alleviates mugwort pollen-induced allergic airway responses through gut microbiota modulation in a murine model. Foods 12(10):2049
Pang W, Jiang Y, Li A, Zhang J, Chen M, Hu L, Li Z, Wang D (2021) Bacteroides thetaiotaomicron ameliorates experimental allergic airway inflammation via activation of ICOS+Tregs and inhibition of Th2 response. Front Immunol 17(12):620943
Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y et al (2011) Induction of colonic regulatory T cells by indigenous Clostridium species. Science 331(6015):337–341
Akagawa S, Kaneko K (2022) Gut microbiota and allergic diseases in children. Allergol Int 71(3):301–309
Spacova I, Van Beeck W, Seys S, Devos F, Vanoirbeek J, Vanderleyden J, Ceuppens J, Petrova M, Lebeer S (2020) Lactobacillus rhamnosus probiotic prevents airway function deterioration and promotes gut microbiome resilience in a murine asthma model. Gut Microbes 11(6):1729–1744
Kamada N, Chen GY, Inohara N, Núñez G (2013) Control of pathogens and pathobionts by the gut microbiota. Nat Immunol 14(7):685–690
Aloisio I, Santini C, Biavati B, Dinelli G, Cencic A, Chingwaru W, Mogna L, Di Gioia D (2012) Characterization of Bifidobacterium spp. strains for the treatment of enteric disorders in newborns. App Microbiol Biotechnol. 96(6):1561–1576
Simone M, Gozzoli C, Quartieri A, Mazzola G, Di Gioia D, Amaretti A, Raimondi S, Rossi M (2014) The probiotic Bifidobacterium breve B632 inhibited the growth of Enterobacteriaceae within colicky infant microbiota cultures. BioMed Res Int 2014:301053
Low JSY, Soh SE, Lee YK, Kwek KYC, Holbrook JD, Van der Beek EM, Shek LP, Goh AEN, Teoh OH, Godfrey KM et al (2017) Ratio of Klebsiella/Bifidobacterium in early life correlates with later development of paediatric allergy. Benef Microbes 8(5):681–695
Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN et al (2010) Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8(3):292–300
De Weerth C, Fuentes S, Puylaert P, De Vos WM (2013) Intestinal microbiota of infants with colic: development and specific signatures. Pediatrics 131(2):e550–e558
Federici S, Kredo-Russo S, Valdés-Mas R, Kviatcovsky D, Weinstock E, Matiuhin Y, Silberberg Y, Atarashi K, Furuichi M, Oka A et al (2022) Targeted suppression of human IBD-associated gut microbiota commensals by phage consortia for treatment of intestinal inflammation. Cell 185(16):2879-2898.e24
Adlerberth I, Wold AE (2009) Establishment of the gut microbiota in Western infants. Acta Paediatr 98(2):229–238
Kubota H, Makino H, Gawad A, Kushiro A, Ishikawa E, Sakai T, Akiyama T, Matsuda K, Martin R, Knol J et al (2016) Longitudinal investigation of carriage rates, counts, and genotypes of toxigenic clostridium difficile in early infancy. Appl Environ Microbiol 82(19):5806–5814
Stephen-Victor E, Chatila TA (2019) Regulation of oral immune tolerance by the microbiome in food allergy. Curr Opin Immunol 60:141–147
Acknowledgements
We thank Majorbio Biological Technology Co., Ltd. for providing technical assistance in this study. We also thank all the parents of the neonates participated in this study.
Funding
This work was supported by the National Natural Science Foundation of China (Grant numbers 81972221 and 81730102).
Author information
Authors and Affiliations
Contributions
The authors’ responsibilities were as follows: RY, YG, and HQ designed the research; RY, HZ, JW, YG, XW, YZ, and LH collected the subjects’ information and samples, analyzed the data, and interpreted the results; YG and RY wrote the manuscript; RY had primary responsibility for the final content and is the guarantor of the contents of this article and this work. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests in this section.
Ethical Approval
This study was performed in line with the principles of the Declaration of Helsinki. Approval was granted by the Ethics Committee of Shanghai Tenth People’s Hospital (Date/No. SHSY-IEC-4.1/21–188/01).
Informed Consent
Informed consent was obtained from all subjects and/or their legal guardian(s).
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
Cite this article
Gong, Y., Zhong, H., Wang, J. et al. Effect of Probiotic Supplementation on the Gut Microbiota Composition of Infants Delivered by Cesarean Section: An Exploratory, Randomized, Open-label, Parallel-controlled Trial. Curr Microbiol 80, 341 (2023). https://doi.org/10.1007/s00284-023-03444-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-023-03444-4