
Abstract
Enterococcus faecalis (E. faecalis) belongs to lactic acid bacteria which can be used as a probiotic additive and feed, bringing practical value to the health of humans and animals. The prebiotic function of tea polyphenols lays a foundation for green tea polyphenols (GTP) to repair the adverse changes of E. faecalis under stress conditions. In this study, RNA-sequence analysis was used to explore the protective effect of GTP on E. faecalis under bile salt stress. A total of 50 genes were found to respond to GTP under bile salts stress, containing 18 up-regulated and 32 down-regulated genes. The results showed that multiple genes associated with cell wall and membrane, transmembrane transport, nucleotide transport and metabolism were significantly differentially expressed (P < 0.05), while GTP intervention can partly alleviate the detrimental effects of bile salt on amino acid metabolism and transport. The present study provides the whole genome transcriptomics of E. faecalis under bile salt stress and GTP intervention which help us understand the growth and mechanism of continuous adaptation of E. faecalis under stress conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Enterococcus faecalis (E. faecalis), a lactic acid bacterium and common inhabitant in the gut, is one of the most common species of Enterococci [1]. E. faecalis strains have been found beneficial to mice and humans by improving the immunity and inhibiting pathogenic infection through producing bacteriocin [2]. In practical applications, some strains are used alone or in combination in livestock and poultry farms to improve animal growth performance, intestinal health, and immune function [3]. E. faecalis plays a positive role in the recovery of hypercholesterolemia mice by regulating cholesterol transport and intestinal microbiota [4]. Meanwhile, they have also been found to be used to treat antibiotic-related diarrhea or irritable bowel syndrome to reduce the host's cholesterol level or improve immunity [5]. E. faecalis may be affected by various environmental factors, such as temperature, pH, and different energy sources, which can trigger changes in its physiological functions and metabolic processes [6]. Bile salts are biological detergents which facilitate fat digestion in the small intestine. Exposure to bile may induce damages in bacterial membrane and nucleic acids, protein misfolding or denaturation as well as osmotic stress [1]. Nevertheless, the potential molecular pathways of E. faecalis exposed to these physiological deleterious impacts remain largely unknown.
Probiotics help maintain the balance of intestinal microbial composition and function, and play a vital role in maintaining the host's health and stability [7]. The existence of probiotics in adversity (specifically reflected in conditions such as acidity and bile salts) can potentially reflect the strength of their viability [8]. There are several enzyme activities in probiotics, such as deglycosylation or decarboxylation, which are all favorable factors that help them survive in adversity [9]. In certain circumstances, polyphenols can be converted into biological forms that are more effective than the original phenolic compounds [10]. Research has demonstrated that green tea polyphenols (GTP) treated with Paecilomyces variotti tannase exhibited a significantly increased antioxidant capacity [11]. Another research found that the addition of GTP increased the tolerance of Pseudomonas aeruginosa to a series of stress conditions such as oxidants, organic acids, and heat [12]. Additionally, phenolic compounds can be used as substrates for probiotics to improve their survivability and function in the gastrointestinal environment [13].
GTP has a wide range of biological activities, and has become a research hotspot. In our previous experiment, GTP showed the potential to enhance ethanol tolerance of Saccharomyces cerevisiae [14], and it partially alleviated the adverse changes related to cell wall, cell membrane and basic metabolism during ethanol stress. Polyphenols have been reported to have the effect of selectively promoting growth of microbiota [9]. Polyphenol compounds may exert their health effects through a mechanism related to their capability to produce an adaptive response at the cellular level, which involves interaction with certain key proteins in activating cell signaling pathways of exposure to environmental stress [7]. GTP can positively improve the imbalance of intestinal microbiota caused by high-fat diet, and significantly reduce the ratio of Firmicutes/Bacteroidetes [15].
RNA sequencing (RNA-seq) can identify functional genes and related metabolic pathways that affect the growth of E. faecalis under the intervention of external conditions through deep sequencing technology [16]. In the adversity environment, the phenotypic characteristics of E. faecalis and the related expression of different genes have undergone significant changes [17]. In our present research, RNA-Seq was used to investigate the genome-wide transcription of E. faecalis which is under bile salt stress with or without GTP intervention. The results may enable us to preferably understand the interaction between E. faecalis and GTP at the molecular level under bile salt stress in vivo. This provides a more reliable theoretical basis for whether GTP is suitable as a prebiotic or potential functional additive for E. faecalis.
Materials and Methods
Chemicals and Reagents
Green tea was obtained locally from Zhejiang province, Ningbo Beilun District, Baifeng tea plantation, and it was harvested in spring, 2021. Polyamide resin was bought from Ocean Chemical Co., Ltd. (Qingdao, China). Other reagents were purchased from Sinopharm Group Co., Ltd. (Shanghai, China). All chemicals and reagents were analytical grade.
Preparation of GTP
Employed a grinder to grind the dried green tea leaves into a powder state, and then passed through a 60-mesh sieve. On the basis of our previous experiment, 1000 g of tea powders were extracted with 16 L of distilled water at 96 °C for 40 min. After extraction, the extract was centrifuged at 4500 g for 15 min, and then the above operations were repeated to treat the insoluble residue. The obtained supernatant is combined and concentrated, and the residue is dissolved, filtered and added to the polyamide column. 80% ethanol was used to elute the polyester amide column, and the elution was monitored and collected automatically by measuring the absorbance at 280 nm. The eluted fractions were analyzed by high-performance liquid chromatography, the desired fractions were collected and concentrated on the polyamide column rely on our previous methods [14], and GTP was obtained by lyophilizing the fractions in a freeze-dry system.
Culture Conditions of E. faecalis 131-2
E. faecalis 131-2 was acquired from China General Microbiological Culture Collection Center, Beijing, China. The strain of E. faecalis 131-2 was originally isolated from infant gut. The activated E. faecalis 131-2 were cultured in MRS broth at 37 °C for 12 h. MRS medium was used for pre-cultivation at 30 °C with aeration and agita-tion (150 rpm). The activated E. faecalis cells were inoculated in 50 mL of fresh MRS medium at 30 °C with an initial OD600 of 0.1. Then 0.5 mL pre-cultured E. faecalis 131-2 was inoculated in 50 mL of MRS medium and incubated at 30 °C in an orbital shaker at 150 rpm till the mid-exponential phase. Bile salts was added into the experimental cultures to a final bile salts con-centration of 10% (v/v) at exponential phase after cultivated for 8 h. They were then randomly divided into four groups. According to our pre-experiment, four kinds of media [MRS broth (natural pH 6.3), pH 6.3 MRS broth with bile salts (0.2%, w/v), pH 6.3 MRS broth with bile salts (0.2%, w/v) and GTP (0.05%, w/v), pH 6.3 MRS broth with GTP (0.05%, w/v)] were prepared and pre-warmed. Then inoculated 1 mL pre-cultured E. faecalis 131-2 into 50 mL above media in order, and divided them into four groups: control group, bile salts group, bile salts-GTP group and GTP group. The E. faecalis 131-2 cells were cultured in incubator at 37 °C and monitored by measuring the optical density of the culture medium at 600 nm (OD600). Cells for scanning electron microscopy (SEM) analysis and RNA-seq analysis were collected after 8 h cultivation [18]. All samples were stored at − 80 °C before use.
SEM Analysis
E. faecalis 131-2 cells from each culture were collected by centrifugation at 5000 g, 4 °C for 8 min. And then they were washed 3 times with 0.9% physiological saline. After those cells were resuspended in 2.5% glutaraldehyde at 4 °C for 4 h and washed 3 times with 0.1 M PBS for 15 min each time. The cells then were rinsed and dehydrated with progressively higher concentrations of alcohol. The cells were eluted with a series of ethanol solutions (30, 50, 70, 80, 90, 95, and 100%) for 10 min per elution, and afterwards with a gradient of tert-butanol and anhydrous ethanol mix mixtures (ratio 1:3, 1:1, 3:1, and 3:0) for 10 min in each mixture [19]. Subsequently, the bacterial cells were lyophilized. Finally, they were coated with a 40:60 gold/palladium alloy and observed with a SEM system (Hitachi S3400N).
Library Construction, RNA Preparation and Sequencing
E. faecalis 131-2 cells from each culture were collected by centrifugation at 5000 g, 4 °C for 8 min. Three independent biological replicates were performed in each treatment. Total RNA was extracted using the TRIzol (Invitrogen, Carlsbad, USA). After extraction, rRNA was removed by Ribo-Zero Magnetic kit (EpiCentre, Wisconsin, USA). In our experiment, Illumina Truseqt™ RNA sample prep Kit was used to construct the library, but dUTP was used to replace dTTP in the dNTP reagent for the synthesis of the second strand of cDNA, so that the base in the second strand of cDNA contained A/U/C/G. Before PCR amplification, the second strand of UNG enzyme cDNA was digested to make the library contain only the first strand of cDNA. Eventually, the sequencing library was obtained by PCR amplification. The final library was tested and sorted by Illumina Hiseq 4000.
Reads Mapping, Annotation and Analysis
Using Illumina paired-end RNA-seq method, we sequenced the transcriptome and generated billions of reads in a single run. By using statistical method, the base distribution and quality fluctuation of each circle of all sequenced reads can be counted. They can also directly reflect the sequencing quality and library construction quality of samples. So as to ensure the accuracy of follow-up biological information analysis, the original sequencing data were filtered to get high-quality clean data. To ensure the smooth progress of subsequent analysis, the specific operation processes are as follows: remove the adapter sequence in reads and reads not inserted in fragments. Cut off the base with lower quality (mass value < 20) at the end of the sequence (3′end). If there are still bases with mass value < 10 in the remaining sequence, remove the whole sequence, otherwise keep it. Remove reads with N ratio over 10%. Discard adapters and sequences with a length less than 75 bp after quality trimming. Genes with ratio changed more than twofold (P < 0.05) were identified to have significantly different gene expression. The ratios were expressed including the up- and down-fold change. Descriptions and annotations of genes were available in the Genome Database of E. faecalis (https://www.ncbi.nlm.nih.gov/genome/?term=Enterococcus+faecalis).
The pathway tool was then used to predict biochemical pathways using the annotation of the E. faecalis 131-2 genes. KEGG pathways and GO terms were retrieved from the KEGG and GO databases.
Definition and Analysis of Differentially Expressed Genes
Read per Kilobase of Transcript per Million Reads Mapped was used to estimate gene expression levels. Through the evaluation of the correlation of sample repetition, we compared the gene expression levels in different libraries. Fragments per kilobase of exon model per Million mapped reads was used to normalize and calculate the transcript levels of expressed genes. Under the condition of stress, the differentially expressed genes (DEGs) were screened by the joint standard of at least twice change and P-value less than 0.05.
Quantitative Real-Time PCR Validation
In order to ensure the reliability of transcriptome results, we selected 10 representative DEGs for real-time quantitative PCR quantitative analysis (Table S1). Total RNA of E. faecalis 131-2 was reverse-transcribed into cDNA by Fastquant RT Kit (with gDNase) (TIANGEN BIOTECH). Then PCR reactions were performed with a LightCycler96® (Roche Diagnostics GmbH, Mannheim, Germany) thermocycler following the procedures: 94 °C for 3 min, followed by 40 cycles of 95 °C for 3 s, 60 °C for 25 s, and a final extension of 72 °C for 2 min. The relative expression level of target genes was measured with the 2−ΔΔCT method.
Statistical Analysis
The data was analyzed by SPSS (SPSS Inc., Chicago, IL, USA, v 17.0.0). One-way analysis of variance (ANOVA) and the post hoc Tukey test were used for comparison between groups. P-value < 0.05 was considered statistically significant.
Results
Effects of GTP on the Growth of E. faecalis
As shown in Fig. 1, compared with the control group, the bile salt group has a stronger inhibitory effect on the growth of E. faecalis 131-2, and GTP supplementation can effectively alleviate the inhibitory effect of bile salt stress on the growth of E. faecalis. This showed that even if bile salt will negatively affect the growth of E. faecalis, the supplementation of GTP will make its growth tend to be normal. And in the absence of bile salt stress, the addition of GTP has no significant effect on the growth of E. faecalis 131-2, indicating that bile salts may one of the factors threaten the normal growth of E. faecalis 131-2.

Effects of GTP on the growth of E. faecalis. Results were obtained from three independent experiment and expressed as mean ± SD (n = 3). *P < 0.05 (Color figure online)
Comparison of the Physiological Phenotypes of E. faecalis 131-2
In the present study, the growth of E. faecalis 131-2 cells were strongly inhibited by bile salt at the exponential phase (P < 0.05), and GTP effectively alleviated the stress induced by bile salt (P < 0.05). It exhibited different cell morphology of E. faecalis 131-2 among the control group (Fig. 2A), the bile salt group (Fig. 2B), and the bile salt-GTP group (Fig. 2C). In the bile salt group, the surface of E. faecalis 131-2 cells were shrunken and certain flocculation was obvious. This may be related to changes in cell surface properties. However, for the bile salt-GTP group, the surface of E. faecalis 131-2 cells were smoother and less flocculation was observed, which indicated that the supplementation of GTP will make its growth tend to be normal and greatly alleviated the adverse consequences induced by bile salt stress. We also observed that the cell surface morphology of E. faecalis 131-2 in the GTP group was very similar to that of the control group and the bile salt group, with no significant negative effects (Fig. 2D).

The electron micrographs of E. faecalis 131-2 from scanning electron microscopy. A (control group), B (bile salt group), C (bile salt-GTP group) and D (GTP group)
Illumina Sequence Data
Libraries representing each sample transcriptome were constructed for RNA-seq analysis. A total of 30.1 GB with clean reads were obtained. The mapping Ratio statistics table as showed in Table S2. The average mass value of over 94% of the total bases was greater than 30. The reference genome of E. faecalis 131-2 was compared and located by using Penguin software. The results showed that a significant proportion (over 83%) of reads in each library matched the E. faecalis 131-2 gene.
Defining DEGs
We analyzed the gene expression difference of the bile salt group and control group, the bile salt group and bile salt-GTP group respectively. The log2FC cutoff used for up-regulated and down-regulated genes here is |log2FC|≥ 1 (Log2FC represents the ratio of expression levels between 2 groups). Compared with the control group, under bile salt stress, E. faecalis 131-2 identified a total of 1169 DEGs, including 588 up-regulated genes and 581 down-regulated genes (Fig. 3). 50 DEGs were identified in the bile salt-GTP group and the bile salt group, containing 18 up-regulated and 32 down-regulated genes. Tables S3-S4 exhibited the DEGs between the bile salt group and the control group, the bile salt-GTP group and the bile salt group, separately.

Screening and expression profiling of DEGs by using volcano plots between the control and bile salt group (A), and bile salt-GTP and bile salt group (B) (Color figure online)
GO Classification Analysis
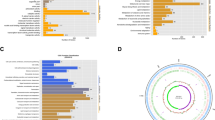
GO classification analysis were divided into three different functional sub-groups to help us better understand the functional classification of DEGs under bile salt stress. As shown in Fig. S1, under bile salt stress compared with the control group abundant GO terms in biological process were participated in “biological regulation”, “cellular process”, “metabolic process”, “multi-organism process”, and “response to stimulus”. Figure 4 exhibited the DEGs analysis of GO enrichment in bile salt-GTP group and bile salt group. After the supplementation of GTP under the bile salt stress, GO terms enriched in terms of biological process were distributed in “biological regulation”, “cellular process” and “metabolic process”. GO analysis of DEGs between the bile salt group and the control group was exhibited in Table S5, and GO analysis of DEGs between the bile salt-GTP group and the bile salt group was exhibited in Table S6. All genes between the bile salt-GTP group and the bile salt group was exhibited in Table S7.

GO analysis of DEGs between the bile salt-GTP group and the bile salt group. Red bars represent up-regulated genes, blue bars represent down-regulated genes (Color figure online)
KEGG Pathway Analysis
Through the enrichment of KEGG pathway and the analysis of DEGs between the bile salt group and the control group, we found sophisticated pathways under bile salt stress. Under the bile salt stress in Fig. S2, the number of genes enriched to "biosynthesis of amino acids" (ko01230, P < 0.05), "aminoacyl-tRNA biosynthesis" (ko00970, P < 0.05), and "oxidative phosphorylation" (ko00190, P < 0.05) is more than that of other functions. Oxidative phosphorylation is a biochemical process that occurs in the mitochondrial inner membrane of eukaryotic cells or in the cytoplasm of prokaryotes. It is the coupling reaction of ADP and inorganic phosphoric acid to produce ATP through the energy released during the oxidation of substances in vivo [20]. In addition, the KEGG pathway "glycolysis/gluconeogenesis" also enriched more differential genes. It has been found that tumor cells have different metabolic changes from normal cells, and tumor cells themselves can adapt to the changes of metabolic environment through the conversion between glycolysis and oxidative phosphorylation [21]. These results suggested that the functions of these KEGG pathways were significantly altered after bile salt stress. As shown in Fig. 5, DEGs were notably enriched in pathway “valine, leucine and isoleucine degradation” (ko00280, P < 0.01) between the bile salt-GTP group and the bile acid group. KEGG analysis of DEGs between the bile salt group and the control group was exhibited in Table S8, while KEGG analysis of DEGs between the bile salt-GTP group and the bile salt group was exhibited in Table S9.

KEGG analysis of DEGs between the bile salt-GTP group and the bile salt group. The diameter of the circle is proportional the number of DEGs enriched in each pathway. The color of circle represents the P-value for enrichment. Rich factor (%) is the ratio of the number of differentially expressed genes annotated in a pathway (as indicated in the y-axis) to the number of all genes annotated in this pathway (Color figure online)
RNA-Seq Expression Validation by Quantitative Real-Time PCR
To quantitatively determine the reliability of the RNA-seq sequencing, we detected the relative expression of 10 randomly selected DEGs by qRT-PCR (Fig. S3). The candidate DEGs contained 5 up-regulated genes and 5 down-regulated genes from Table S10. The data results showed that the gene EF0017, EF0187, EF0368, EF0744, and EF1182 showed an upward trend after GTP intervention. The genes EF0039, EF0121, EF0252, EF0836, and EF1166 showed a downward trend. RNA-seq and RT-qPCR data were highly consistent, manifesting the reliability of transcriptome results.
Discussion
The main component of the Enterococcus cell wall is peptidoglycan, which plays a vital role in maintaining the shape of bacterial cells and resisting internal and external pressure. [8]. Bacterial surface proteins compose a special molecular library with many important functions, such as adhesion, invasion, signaling, and interaction with host immune system or environment [22]. The significant differential expression of genes may be due to compensation mechanisms to counteract bile salt stress and protect the bacterial cell wall from environment damage. Under bile salt stress, the genes EF0093 for LPXTG-motif cell wall anchor domain protein and EF3314 for cell wall surface anchor protein were up-regulated, while EF2685 for LPXTG cell wall anchor domain-containing protein, EF2486 for teichoic acids export ATP-binding protein TagH was down-regulated.
Bacterial cell membrane acts as an indispensable role in the selective absorption of nutrients, the discharge of wastes, and the adaptation and response of external environment [23]. Membrane proteins are abundant in cells, which included signal and recognition receptors, ion transport proteins and channels, structural proteins and enzymes [24]. In our experiment under bile salt stress, we discovered that genes EF0025, EF0397, EF0943, EF1189, EF1288, EF2383, EF2771 to encode membrane protein were down-regulated. These results indicate that membrane proteins participated in and were affected by the stress response of E. faecalis against external conditions [25]. Wang et al. reported that 3-Phenyllactic acid can change the permeability of E. faecalis cell membrane and cause the leakage of intracellular components [26]. There was a study showed that bile salts can damage the cell membrane of bacteria, and change the phenotype of cells by affecting certain biological processes (e.g., protein denaturation, the composition of fatty acids and phospholipids) [27]. We found genes EF0991 for penicillin-binding protein, EF0992 for phosphor-N-acetylmuramoyl-pentapeptide transferase, EF2489 for UDP-N-acetylmuramate dehydrogenase, and EF0992 for UDP-N-acetylmuramoyl-L-alanine-D-glutamate ligase participated in peptidoglycan biosynthesis were also prominently down-regulated. 10 genes (EF0282-0284, EF0365, EF1667, EF2875-2878, EF2880) participated in fatty acid biosynthesis were also observed the same decline. After GTP intervention under the bile salt stress, we found that the expression of genes related to E. faecalis cell wall and cell membrane did not alter significantly under bile salt stress. This may signify that the cell wall and membrane of E. faecalis were not the main action site of GTP, as well as not injured by GTP.
The heat shock protein DnaK is a molecular chaperone protein that participates in a variety of cellular activities. DnaK itself has a low endogenous rate of ATPase, which may accelerate modulation of the vital co-chaperone GrpE [28]. Under bile salt stress, two genes EF1307 for nucleotide exchange factor GrpE and EF1308 for molecular chaperone DnaK which encoding the two common heat shock proteins were up-regulated. This may imply the potential self-protection mechanism of E. faecalis under stress. Universal stress proteins are one of the most abundant proteins in growth-restricted cells, so the down-regulated genes may indicate that bile salt stress still has a negative impact on cells to a certain extent [29]. Study has reported that the Gls24 superfamily proteins were involved in the survival of pathogenic Gram-positive cocci under oligotrophic conditions and other types of stress conditions through molecular mechanisms [30]. And Gls24 was previously considered to be a common stress protein of E. faecalis. Inactivation of Gls24 will lead to a decrease in the anti-bile salt stress capacity of E. faecalis [31]. However, genes EF1058 encoding universal stress protein and EF3115 encoding Asp23/Gls24 family envelope stress response protein were down-regulated under bile salt stress. No obvious changes were found after adding GTP.
Amino acids are the main metabolites in the biosynthesis and metabolism of E. faecalis [32]. Metabolites produced by the decomposition of amino acids follow the metabolic pathway of sugar or lipid with different characteristics, and release energy ultimately [33]. Previous study has shown that some amino acids can be used as energy sources by E. faecalis [34]. Under bile salt stress, more than 20 genes (such as EF0043, EF0045, EF0105, EF0246, EF0451 etc.) associated with amino acid transport and metabolisms were significantly down-regulated. This indicated that acidic stress may affect the transportation and supply of amino acids, thereby inhibiting the growth and reproduction of E. faecalis. While interestingly, compared with the bile salt group, the genes EF0105 and EF0805 which were related to amino acid transport and metabolism were significantly up-regulated after GTP intervention. This revealed that GTP intervention may partially alleviate the negative effects of bile salt on amino acid transport and metabolism. Genes involved in nucleotide transport and metabolism show differential expression under bile acid stress. The genes (EF0688, EF0689, EF0807, EF0862, EF0864, EF0911, and EF0941) which were related to “ABC transporter, ATP-binding protein” and “ribonucleotide reductase assembly protein Nrdl” showed significant down-regulation under bile salt stress. This may indicate that E. faecalis is inhibited in internal nucleotide transport and metabolism under the stress of bile acid. The good survival conditions of E. faecalis not only depend on the protection of the genome, but also on the balance of the external environment, so that it cannot be affected by changes that lead to cell pathology and reduced adaptability.
The 16 related genes (EF0019-EF0022, EF0516, EF0717, EF1013, EF1016-EF1019, EF1359, EF1769, EF1829, EF1830, EF1838, and EF2603) encoding the phosphotransferase system (PTS) were significantly down-regulated under bile salt stress. The results showed that they were involved in the mannose, fructose, sorbose family and PTS sugar transporter subunit IIABCD processes in the PTS system to varying degrees. The protein of sugar PTS can not only catalyze the uptake of carbohydrates, but also modulate the activity of multiple genes and proteins to make better use of available carbon sources [35]. A key factor of the modulation of carbohydrate metabolism is the PTS. The term “PTS” refers to a group of enzymes that can transfer the phosphate moiety from phosphoenolpyruvate (PEP) from one part of the system to the next in a relatively fixed order [36]. PEP contained carbohydrate PTS, or sugar PTS, which was mainly responsible for the accompanying transport and phosphorylation of carbohydrates into cells [37]. PTS has been found in different types of bacteria, and there were differences in order to better evolve to find the most suitable substrate carbon source [36]. After supplementing of GTP, 11 genes (EF0019-EF0022, EF0516, EF0717, EF1013, EF1019, EF1359, EF1769, EF2603) were up-regulated among the above-mentioned 16 genes of down-regulated. This indicated that under bile acid stress, substrates other than glucose may also contribute to the growth of E. faecalis.
Furthermore, the gene EF0718 which encoded 1-phosphofructokinase, the gene EF1049 which was involved in NADP-dependent phosphogluconate dehydrogenase, and the gene EF2425 encoding phospho-sugar mutase, reversed the downward trend of these genes in E. faecalis after GTP intervention. This manifested that the glycolytic pathway of E. faecalis was inhibited in the bile salt environment, and the bacteria were beginning to choose non-preferred carbon and energy sources (such as various types of sugars), and GTP would assist this process.
ATP supports almost all cellular activities that need energy. ATP synthesis is the most common chemical reaction in the biological world. ATPase is one of the most common and abundant proteins on earth [38]. Under the bile salt stress, eight genes (EF2607, EF2608, EF2609, EF2610, EF2611, EF2612, EF2613, EF2614) participated in ATP synthase were down-regulated. In addition, the gene EF2623 involved in cadmium-translocating P-type ATPase which can couple ATP hydrolysis to the translocation of protons across bacterial cell membranes was also down-regulated. However, compared with the bile salt environment, the genes (EF0805, EF1505, EF1674, EF1918) which respectively belong to amino acid ABC transporter ATP-binding protein, alpha/beta hydrolase, hypothetical protein and hypothetical protein EFDM72_1381 was significantly up-regulated in the bile salt-GTP group. It showed that the physiological functions related to ATP in E. faecalis may be weakened in the bile salt environment, and GTP may partially improve this phenomenon.
Conclusions
In this study, we mainly used RNA-seq analysis to reveal the corresponding changes of E. faecalis 131-2 cells under bile salt stress. It also revealed the effects of GTP on E. faecalis under bile salt stress. The most enriched metabolic pathway of DEGs in the bile salt-GTP group compared with the bile salt group were valine, leucine and isoleucine degradation, selenocompound metabolism, and PTS. These results may provide a more directional reference for further research on the effect of GTP on E. faecalis under bile salt environments in the future. The interaction mechanism between GTP and E. faecalis needs to be further explored in order to promote the application of E. faecalis and GTP in fermented foods and probiotic supplements.
Data Availability
The authors confirm that all data generated or analyzed during this study are included in this published article [and its supplementary information files]. The NCBI BioProject is PRJNA796179 [Transcriptome sequencing of Enterococcus faecalis].
Abbreviations
- E. faecalis :
-
Enterococcus faecalis
- E. faecalis 131-2:
-
Enterococcus faecalis131-2
- GTP:
-
Green tea polyphenols
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- SEM:
-
Scanning electron microscopy
- DEGs:
-
Differentially expressed genes
References
Salze M, Giard JC, Riboulet BE, Hain T, Cécile M (2019) Identification of the general stress stimulon related to colonization in Enterococcus faecalis. Arch Microbiol 202:233–246
Li P, Niu Q, Wei Q, Zhang Y, Ma X, Kim SW, Lin M, Huang R (2017) Microbial shifts in the porcine distal gut in response to diets supplemented with Enterococcus faecalis as alternatives to antibiotics. Sci Rep 7:1–10
Shehata AA, Tarabees R, Basiouni S, Elsayed MS, Gaballah A, Krueger M (2020) Effect of a potential probiotic candidate Enterococcus faecalis-1 on growth performance, intestinal microbiota, and immune response of commercial broiler chickens. Probiotics Antimicro 12:451–460
Zhu Y, Li T, Din AU, Hassan A, Wang Y, Wang G (2019) Beneficial effects of Enterococcus faecalis in hypercholesterolemic mice on cholesterol transportation and gut microbiota. Appl Microbiol Biotechnol 103:3181–3191
Franz CM, Huch M, Abriouel H, Holzapfel W, Gálvez A (2011) Enterococci as probiotics and their implications in food safety. Int J Food Microbiol 151:125–140
Lindenstrauß AG, Ehrmann MA, Behr J, Landstorfer R, Haller D, Sartor RB, Vogel RF (2014) Transcriptome analysis of Enterococcus faecalis toward its adaption to surviving in the mouse intestinal tract. Arch Microbiol 196:423–433
Monagas M, Urpi SM, Sánchez PF, Llorach R, Garrido I, Gómez C, Andres C, Bartolomé B (2010) Insights into the metabolism and microbial biotransformation of dietary flavan-3-ols and the bioactivity of their metabolites. Food Funct 1:233–253
İspirli H, Demirbaş F, Dertli E (2015) Characterization of functional properties of Enterococcus faecium strains isolated from human gut. Can J Microbiol 61:861–870
Banerjee A, Dhar P (2019) Amalgamation of polyphenols and probiotics induce health promotion. Crit Rev Food Sci Nutr 59:2903–2926
López de Lacey AM, Pérez-Santín E, López-Caballero ME, Montero P (2014) Biotransformation and resulting biological properties of green tea polyphenols produced by probiotic bacteria. LWT Food Sci Technol 58:633–638
Macedo JA, Battestin V, Ribeiro ML, Macedo GA (2011) Increasing the antioxidant power of tea extracts by biotransformation of polyphenols. Food Chem 126:491–497
Liu X, Li J, Yang Y, Chen X (2012) Exposure of Pseudomonas aeruginosa to green tea polyphenols enhances the tolerance to various environmental stresses. World J Microbiol Biotechnol 28:3373–3380
De Souza EL, De Albuquerque TMR, Dos Santos AS, Massa NML, de Brito Alves JL (2019) Potential interactions among phenolic compounds and probiotics for mutual boosting of their health-promoting properties and food functionalities-a review. Crit Rev Food Sci Nutr 59:1645–1659
Cheng L, Zhang X, Zheng XJ, Wu ZF, Weng PF (2019) RNA-seq transcriptomic analysis of green tea polyphenols regulation of differently expressed genes in Saccharomyces cerevisiae under ethanol stress. World J Microbiol Biotechnol 35:59
Guo XJ, Cheng M, Zhang X, Cao JX, Wu ZF, Weng PF (2017) Green tea polyphenols reduce obesity in high-fat diet-induced mice by modulating intestinal microbiota composition. Int J Food Sci Tech 52:1723–1730
Zhang XL, de Maat Vd, Prieto AMG, Prajsnar TK, Bayjanov JR, Been Md, Rogers MRC, Bonten MJM, Mesnage S, Willems RJL (2017) RNA-seq and Tn-seq reveal fitness determinants of vancomycin-resistant Enterococcus faecium during growth in human serum. BMC Genomics 18:893
Solheim M, La Rosa SLL, Mathisen T, Snipen LG, Nes IF, Brede DA (2014) Transcriptomic and functional analysis of NaCl-induced stress in Enterococcus faecalis. PLoS ONE 9:e94571
Lee JY, Pajarillo AB, Kim MJ, Chae JP, Kang DK (2012) Proteomic and transcriptional analysis of Lactobacillus johnsonii PF01 during bile salt exposure by iTRAQ shotgun proteomics and quantitative RT-PCR. J Proteome Res 12:432–443
Chen YH, Cheng L, Zhang X, Cao JX, Wu ZF, Zheng XJ (2019) Transcriptomic and proteomic effects of (-)-epigallocatechin 3-O-(3-O-methyl) gallate (EGCG3”Me) treatment on ethanol-stressed Saccharomyces cerevisiae cells. Food Res Int 119:67–75
Zheng XD, Boyer L, Jin MJ, Mertens J, Kim Y, Ma L, Hamm M, Gage FH, Hunter T (2016) Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. Elife 5:e13374
Elgendy M, Cirò M, Hosseini A, Weiszmann J, Mazzarella L, Ferrari E, Cazzoli R, Curigliano G, DeCensi A, Bonanni B, Budillon A, Pelicci PG, Janssens V, Ogris M, Baccarini M, Lanfrancone L, Weckwerth W, Foiani M, Minucci S (2019) Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3β-MCL-1 axis. Cancer Cell 35(5):798-815.e5
Dramsi S, Bierne H (2017) Spatial organization of cell wall-anchored proteins at the surface of gram-positive bacteria. Curr Top Microbiol Immunol 404:177–201
Silhavy TJ, Kahne D, Walker S (2010) The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414
Foot N, Henshall T, Kumar S (2017) Ubiquitination and the regulation of membrane proteins. Physiol Rev 97:253–281
Giard JC, Laplace JM, Rincé A, Pichereau V, Benachour A, Leboeuf C, Flahaut S, Auffray Y, Hartke A (2001) The stress proteome of Enterococcus faecalis. Electrophoresis 22:2947–2954
Wang F, Wu H, Jin P, Sun Z, Liu F, Du L, Wang D, Xu W (2018) Antimicrobial activity of phenyllactic acid against Enterococcus faecalis and its effect on cell membrane. Foodborne Pathog Dis 15:645–652
Urdaneta V, Casadesús J (2017) Interactions between bacteria and bile salts in the gastrointestinal and hepatobiliary tracts. Front Med 4:163
Chang L, Thompson AD, Ung P, Carlson HA, Gestwicki JE (2010) Mutagenesis reveals the complex relationships between ATPase rate and the chaperone activities of Escherichia coli heat shock protein 70 (Hsp70/DnaK). J Biol Chem 285:21282–21291
Kvint K, Nachin L, Diez A, Nyström T (2003) The bacterial universal stress protein: function and regulation. Curr Opin Microbiol 6:140–145
Ferrándiz MJ, Cercenado MI, Domenech M, Tirado-Vélez JM, Escolano-Martínez MS, Yuste J, García E, de la Campa AG, Martín-Galiano AJ (2019) An uncharacterized member of the Gls24 protein superfamily is a putative sensor of essential amino acid availability in Streptococcus pneumoniae. Microb Ecol 77:471–487
Teng F, Nannini EC, Murray BE (2005) Importance of gls24 in virulence and stress response of Enterococcus faecalis and use of the Gls24 protein as a possible immunotherapy target. J Infect Dis 191:472–480
Paddon-Jones D, Rasmussen BB (2009) Dietary protein recommendations and the prevention of sarcopenia protein, amino acid metabolism and therapy. Curr Opin Clin Nutr Metab Care 12:86–90
Dawczynski C, Schubert R, Jahreis G (2007) Amino acids, fatty acids, and dietary fibre in edible seaweed products. Food Chem 103:891–899
Alomar J, Loubière P, Delbes C, Nouaille S, Montel MC (2008) Effect of Lactococcus garvieae, Lactococcus lactis and Enterococcus faecalis on the behaviour of Staphylococcus aureus in microfiltered milk. Food Microbiol 25:502–508
Deutscher J, Francke C, Postma PW (2006) How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031
Pflüger-Grau K, Görke B (2010) Regulatory roles of the bacterial nitrogen-related phosphotransferase system. Trends Microbiol 18:205–214
Postma PW, Lengeler JW, Jacobson JR (1993) Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57:543–594
Yoshida M, Muneyuki E, Hisabori T (2001) ATP synthase-a marvellous rotary engine of the cell. Nat Rev Mol Cell Biol 2:669–677
Funding
This work was supported by the Fundamental Research Funds for the Central Universities (2020SKTY01), the National Natural Science Foundation of China (31650006), "Yueqi Young Scholars" Funding Program of China University of Mining and Technology (Beijing), and the Key Research and Development Project of Zhejiang Province (2018C02047).
Author information
Authors and Affiliations
Contributions
LZ and JX drafted the manuscript. LZ and ZZ analyzed the RNA sequencing data. LZ, ZW, and RY conducted the statistics study. All authors critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflict of interest.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.



Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Zhang, L., Xie, J., Zhang, Z. et al. RNA-Seq Transcriptomic Analysis of Green Tea Polyphenols Modulation of Differently Expressed Genes in Enterococcus faecalis Under Bile Salt Stress. Curr Microbiol 79, 147 (2022). https://doi.org/10.1007/s00284-022-02844-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00284-022-02844-2