
Abstract
Pseudomonas aeruginosa is a common opportunistic pathogen that causes infections in vulnerable patients including those with metabolic disorders, hematologic diseases, and malignancies, and in those who have undergone surgery. In addition, P. aeruginosa exhibits high intrinsic resistance to numerous antibiotics and tends to form biofilms rendering it even more refractory to treatment. Among the mechanisms used by P. aeruginosa to adapt to environmental stresses are those involving small regulatory RNAs (sRNAs), which are 40–500 nucleotides long and are ubiquitous in bacteria. sRNAs play important regulatory roles in various vital processes in diverse bacteria, with their quantity and diversity of regulatory functions exceeding those of proteins. In this study, we show that deletion of the sRNA, rgsA, decreased the growth rate of P. aeruginosa. Furthermore, ΔrgsA P. aeruginosa exhibited decreased ability to resist the stress induced by exposure to different concentrations and durations of peroxides in both planktonic and biofilm growth modes compared with the wild-type strain. These results highlight the role of rgsA in the defense of P. aeruginosa against oxidative stress.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Aerobic and facultative anaerobic bacteria primarily employ aerobic respiration for energy metabolism. During this process, aberrant electron transport in the electron transport chain or cell oxidoreductase may produce reactive oxygen intermediates (ROIs), such as superoxide, hydrogen peroxide (H2O2), and hydroxyl free radicals [1]. In addition, bacteria may be exposed to exogenous ROIs, particularly during infection of the human body, when human phagocytes (such as neutrophils) exert significant oxygen-dependent antibacterial effects [2, 3]. These ROIs may cause oxidative damage to cellular proteins, cell membranes, and genetic substances, such as DNA [4]. Bacteria possess a series of protective systems to resist oxidative stress, which involve antioxidant proteins (superoxide dismutase, catalase, and peroxidase), iron sequestration, free radical scavengers, DNA-binding proteins, and DNA repair enzymes [5].
Pseudomonas aeruginosa is an opportunistic pathogen that demonstrates high antimicrobial resistance. It also exhibits extremely strong resistance against complex factors, such as immune responses, antibiotics, oxidative stress, and osmotic stress [6]. P. aeruginosa causes about 65% of the chronic infections in humans owing to its ability to form biofilms. Infections, such as dental plaque, cystic fibrosis, recurrent or chronic otitis media, chronic osteomyelitis, chronic sinusitis, and chronic wound infections [7], are related to biofilm formation. Bacteria in biofilms possess a unique gene expression mechanism as compared with their planktonic counterparts [8]. The most important characteristic of the “biofilm phenotype” is its significantly greater tolerance toward changing environments [9]. Therefore, biofilm phenotypes often cause persistent infections leading to protracted diseases [10]. Numerous mechanisms in P. aeruginosa have evolved to respond to specific environmental signals. Among these is the mechanism involving an important class of regulators, the small regulatory RNAs (sRNAs).
sRNAs are 40–500 nucleotides long, possess regulatory functions, and are ubiquitous in bacteria. They play important regulatory roles in various vital life processes in diverse bacteria [11]. Generally, the expression of sRNA molecules is induced by certain transcription factors under specific environmental conditions, metabolic signals, or survival stresses [12]. sRNAs can regulate many targets or act as global regulatory factors and participate in multiple biological processes. sRNA regulation is usually conserved, and the extent and diversity of its regulatory effects exceed those of proteins [13]. In recent years, there have been several reports on sRNAs in P. aeruginosa [14], including PrrF1, PrrF2, and PrrH, which are induced by low iron levels and participate in the regulation of iron homeostasis [15]; RsmY and RsmZ, which are key regulators in the CbrA/CbrB signal transduction pathway and regulate N-butanoyl-homoserine lactone levels, and factors associated with acute and chronic virulence phenotypes [16]; PhrS, which stimulates the synthesis of the P. aeruginosa quinolone signal and affects the production of pyocyanin; and CrcZ, which acts as a mediator of carbon catabolite repression [17].
Many sRNAs play important roles in protective processes. Among them, rgsA has attracted the attention of researchers in recent years. rgsA, which was discovered by whole genome sequencing in 2008, is an sRNA present in Pseudomonas species, including P. aeruginosa, Pseudomonas fluorescens, Pseudomonas putida, and Pseudomonas syringae, with a predicted gene length of 120 bp, produced more than one transcript [18]. In a previous study, it was shown that rgsA accumulates to a peak level when cells are in the stationary growth phase [19]. rgsA expression is directly regulated by the major stationary phase and stress sigma factor σS (RpoS), and it is indirectly regulated by the GacS/GacA two-component system [20]. The expression level of rgsA in biofilms at 48 h of culture was shown to be 1000 times of that in planktonic bacteria at 4 h (exponential phase) and 12 h (stationary phase) [21]. It has been reported that deletion of rgsA may cause a decrease in the resistance of the bacteria to H2O2 [18], but the influence of different oxidative stress conditions on P. aeruginosa in planktonic and biofilm growth modes has not been studied. In this study, we investigated the effects of rgsA deletion on oxidative stress resistance in P. aeruginosa to decipher the mechanism underlying the role of rgsA in oxidative stress resistance.
Materials and Methods
Bacteria, Plasmids, and Growth Conditions
Table 1 contains information on bacterial strains and plasmids used in this study. The culture media used consisted of Luria broth (LB) and nutrient agar plates. For screening of bacteria, 100 μg mL−1 ampicillin, 25 μg mL−1 (Escherichia coli) or 100 μg mL−1 (P. aeruginosa) tetracycline, 10 μg mL−1 (E. coli) or 40 μg mL−1 (P. aeruginosa) gentamycin, and 100 μg mL−1 (E. coli) or 300 μg mL−1 (P. aeruginosa) spectinomycin were used. E. coli and P. aeruginosa were inoculated from glycerol stocks onto nutrient agar plates and cultured for 18–20 h. Following this, a single colony was selected, subcultured in LB, and then cultured to the pre-logarithmic growth phase, which was indicated by an optical density of 0.5 at 600 nm (OD600 ≈ 0.5). The bacterial suspension obtained was centrifuged and the pellet was washed before analyses.
DNA and RNA Extraction
Bacterial genomic DNA was extracted using a QIAquick PCR Purification Kit (Qiagen, Germany). TIANprep Mini Plasmid Kit (Tiangen Biotech, China) was used for plasmid DNA extraction and a QIAquick Gel Extraction Kit (Qiagen) was used for the recovery of plasmids from gels. The total RNA was extracted using the TRIzol method [28] and treated with RNase-free DNase (Promega, USA) to remove any residual genomic DNA. The integrity of the isolated RNA was assessed by agarose gel electrophoresis and the concentration of RNA was determined using a UV spectrophotometer (Eppendorf, Germany). The intensity and sharpness of the 5S, 16S, and 23S bands were used to assess RNA integrity, and A260/A280 values were measured to assess RNA purity. RNA (5 μL), no reverse transcriptase control, and positive PCR control of PAO1 genomic DNA were used as templates in polymerase chain reaction (PCR) amplification (40 cycles) of the 16S rRNA gene to detect any DNA contamination in the RNA samples. The PCR products were visualized after electrophoresis on 1% (wt/vol) agarose gel; the presence of any band of RNA and no RNA control sample were indicative of DNA contamination.
Determination of the rgsA Transcription Start Site
5′-RACE was employed to determine the transcription start site of rgsA [18]. P. aeruginosa PAO1 was cultured in LB medium until an OD600 of 0.5 (pre-logarithmic phase) was reached, and then RNA was extracted from the pelleted bacteria. A cDNA reverse transcription system was used to synthesize first strand cDNA from the extracted RNA. After purification and recovery, terminal deoxynucleotidyl transferase (TdT) and dATP were used to add a poly(A) tail to the 5′-terminus of the cDNA. The product of this reaction was used as a template for RACE. The RACE-anchor-T17 primer containing 17–20 Ts at the 3′-end as well as the specific primers, RgsA-R3 and GM-PA2958-R1, were used for three rounds of nested PCR to amplify the transcripts. After recovery, the amplified DNA fragments were cloned in a T-vector, pMD19-T. The generated constructs were transformed into DH5α competent cells. Next, plasmid was extracted from the positive strains cultured in 10 tubes. Plasmids from six tubes were used for sequencing and alignment with the gene library to determine the transcription start site.
Construction of rgsA Deletion Strains
An overnight culture of P. aeruginosa PAO1 was collected and genomic DNA was extracted. Two pairs of primers (HindIII-PA2957-F1/PA2959-PstI-PA2958-R1 and PA2959-PstI-PA2958-F1/BamHI-PA2960-R3) were used for PCR amplification of the upstream homology arm PA2958 and the downstream homology arm PA2959 of the rgsA gene, respectively. The primers were designed such that the ends of the products, PA2958 and PA2959, contained 47 bp complementary sequences. The mixture of PA2958 and PA2959 was subjected to overlap extension PCR with the primer pair HindIII-PA2957-F1/BamHI-PA2960-R3. After gel purification and digestion with restriction enzymes (HindIII and BamHI), the PCR product PA2958-PA2959 was cloned into a suicide vector, pEX18-Ap. The ligated vector was subsequently transformed into DH5α competent cells, which were screened on ampicillin plates. Plasmids were extracted and digested with PstI, and this was followed by ligation with the gentamicin resistance gene fragment that was obtained after PstI digestion of the pUCGM plasmid. The ligated product was then transformed into competent E. coli SM10 to construct the recombinant E. coli SM10 strain (pEX18-PA2958-PA2959-aacC1), which was screened on plates containing ampicillin and gentamicin. Two-parent hybridization was used to transfer the recombinant plasmid in E. coli SM10 (pEX18-PA2958-PA2959-aacC1) to PAO1. The suicide vector-mediated homologous recombination method was used, and LB medium containing the screening antibiotic and 15% sucrose was employed for screening the PAO1ΔrgsA mutant. DNA was extracted from the screened mutants. The primer pair HindIII-PA2957-F1/BamHI-PA2960-R3 was used for PCR amplification to detect deletions in rgsA. The expected size of the PCR product was 3357 bp.
Construction of the rgsA Overexpression Strain
Based on the results of 5′-RACE, EcoRI-rgsA-F1/PstI-rgsA-R1 was used to amplify the rgsA short transcript fragment. NcoI was used to digest pJN105, which was ligated with the tet gene that was obtained from NcoI digestion of pEX18-TE to obtain the pJNT plasmid that would confer tetracycline resistance. The pJN105 plasmid contained the araC-pBAD control expression cassette, which was induced in the presence of l-arabinose [27]. The pJNT and rgsA short transcript gene fragments were double digested with EcoRI and PstI, and this was followed by ligation to obtain the recombinant plasmid pJR1, which was subsequently transformed into competent E. coli SM10 cells to obtain the E. coli SM10 (pJR1) recombinant strain. The colonies were screened on plates containing a screening antibiotic. Finally, two-parent hybridization was used to transfer the recombinant plasmid in E. coli SM10 (pJR1) to PAO1ΔrgsA to obtain the PAO1ΔrgsA (pJR1) overexpression strain.
Construction of Parallel Experimental Strains
The pJNT plasmid was transformed into competent E. coli SM10 cells to obtain the E. coli SM10 (pJNT) strain. Two-parent hybridization was used to transfer the plasmid in E. coli SM10 (pJNT) to PAO1 and PAO1ΔrgsA to obtain the parallel experimental strains, PAO1 (pJNT) and PAO1ΔrgsA (pJNT), respectively.
Analysis of Growth Curve
A total of 0.5 mL of PAO1 and PAO1ΔrgsA overnight culture was subcultured in 50 mL LB medium and then cultured at 37 °C with shaking at 200 rpm. The OD600 was measured every 15 min. Each measurement was conducted in triplicate and means were calculated after which the values were recorded and used to plot a curve to calculate the specific growth rate and doubling time.
Real-time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
The bacterial strains were grown at 37 °C until an OD600 of 0.5 was reached. Different volumes of H2O2 solution (2.25, 6.65, and 9.0 mM final concentration), cumene hydroperoxide (CHP) (0.5 mM final concentration), and tert-butyl hydroperoxide (TBH) (0.5 mM final concentration) were added separately to culture bottles and shaken for 30 min. Total RNA was subsequently isolated from the strains using TRIzol. First-strand cDNA was produced using random primers and Quant Reverse Transcriptase (Tiangen, Beijing). The levels of rgsA mRNA and the reference gene, 16S rRNA, were quantitatively assessed by real-time RT-PCR using the RealMaster Mix (SYBR Green) (Tiangen, Beijing). Real-time RT-PCR reactions were carried out in a LightCycler® 480 and data were analyzed using the LightCycler® 480 software. The relative rgsA mRNA expression level was calculated using the 2−ΔCt method, as follows: ΔCt (rgsA) = Ct (rgsA) – Ct (16 s rRNA). The data were analyzed using Student’s t test to determine whether the difference was statistically significant. The fold changes in expression were calculated using the 2−ΔΔCt method, as follows: ΔΔCt (rgsA) = ΔCt (rgsA in treatment group) − ΔCt (rgsA in control group) [29].
Plate Survival Experiments
PAO1 (pJNT), PAO1ΔrgsA (pJNT), and PAO1ΔrgsA (pJR1) cultures were separately diluted with 0.85% NaCl + 0.1% l-arabinose solution to an OD600 of 0.3, and this was followed by tenfold serial dilution using the same solution to obtain cultures with six different densities of bacteria. Next, the diluted samples were inoculated on plates containing 2 mM H2O2, 0.5 mM CHP, and 0.5 mM TBH. The plates were incubated for 24 h and bacterial survival was analyzed.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay for Quantification of Cell Viability
PAO1 (pJNT), PAO1ΔrgsA (pJNT), and PAO1ΔrgsA (pJR1) were resuspended in 0.85% NaCl + 0.1% l-arabinose solutions. The concentrations of the suspensions were adjusted to an OD600 of 0.15–0.17, after which they were aliquoted into 1 mL Eppendorf tubes. The bacteria were then exposed to the following conditions for simulation of oxidative stress: 40 mM H2O2 for 30 min, 10 mM H2O2 for 2 h, and 10 mM H2O2 for 5 h. The process was repeated for each sample three times. Thereafter, the cell suspensions were centrifuged, and the pellets were washed. Next, each pellet was resuspended in 100 μL 0.85% NaCl solution, and 10 μL MTT was added to it. The samples were then incubated at 37 °C for 60 min in open tubes and centrifuged at 10,000×g for 1 min, after which 99 μL of supernatant was aspirated, and 1250 μL dimethyl sulfoxide (DMSO) solution was used to dissolve the pellet. Finally, the OD550 value was measured after 10–15 min of incubation, and the ratio of the values for the samples and the negative control was calculated.
In Vitro Biofilm Culture
A flow apparatus was used for biofilm culture. PAO1 (pJNT), PAO1ΔrgsA (pJNT), and PAO1ΔrgsA (pJR1) were separately diluted with 0.85% NaCl + 0.1% l-arabinose solution to a McFarland standard of 0.5. Acid-treated glass slides were then immersed in the prepared bacterial suspension, incubated at 28 °C for 24 h, and then placed in a flow apparatus. The perfusate was 10% LB + 0.1% l-arabinose solution, which was subjected to continuous culture for 72 h at a flow rate of 0.05 mL s−1. Phosphate-buffered saline (PBS) was then added as the perfusate, and planktonic bacteria on the glass slides were flushed for 10 min at a flow rate of 0.5 mL s−1. The perfusion device was then removed, and the glass slides were washed once with PBS and then immersed in 10, 20, or 30 mM H2O2 solution for 30 min each. PBS was subsequently used to repeatedly wash the glass slides, which were slightly dried, and then covered with 1,000-fold diluted SYTO9/PI dye. Staining was conducted for 20 min, after which the glass slides were removed from the solution, excess water was removed, and the slides were kept moist until they were observed under a fluorescence microscope (Olympus, Japan).
Results
Identification of the rgsA Transcription Start Site
Two transcripts of different lengths were present in the 5′-RACE product. DNA fragments recovered from the gel were cloned in the pMD19-T-vector. For each type of transcript, plasmids obtained from six culture tubes were used for sequencing and alignment. Based on the results of sequencing of rgsA and sequence alignment, the transcription start site of the short transcript was found to be 86 bp downstream and that of the long transcript was found to be 157 bp upstream of the start site in the annotated gene fragment present in the NCBI database. The transcription terminator might be common in the two transcripts (Fig. 1).

Determination of the transcription start site (TSS) and sequence of Pseudomonas aeruginosa rgsA by 5′-RACE. The TSS is indicated by + 1
Effect of rgsA Deletion on the Growth Rate of P. aeruginosa
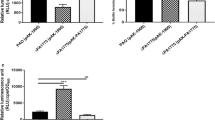
We measured the OD600 values of PAO1 and PAO1ΔrgsA for 10 h to determine whether rgsA affects the growth rate of P. aeruginosa. The growth rate of the wild-type strain was 1.3 times faster than that of the rgsA-deleted mutant strain in the logarithmic phase (Table 2).
Sensitivity of P. aeruginosa Planktonic Cells with Mutations in rgsA to Peroxide and Superoxide Stress
First, the expression levels of rgsA under different oxidative stress conditions were measured using real-time RT-PCR. The strains treated with H2O2 exhibited higher expression levels of rgsA compared with the control strain (Fig. 2).

Real-time RT-PCR data for rgsA expression levels after exposure to different concentrations of hydrogen peroxide solution for 30 min. Data are presented as mean ± SD for three independent experiments. *Marks statistical differences when compared to the control (untreated strain) (Student's t test; P < 0.05)
To investigate the effects of different oxidative stress-inducing substances on the growth of strains containing the rgsA mutant, we conducted plate survival experiments to measure the resistance of PAO1ΔrgsA (pJNT), PAO1ΔrgsA (pJR1), and PAO1 (pJNT) to H2O2 and organic peroxides. Growth inhibition caused by treatment with H2O2, CHP, and TBH was measured. The resistance of strains harboring the rgsA-deleted mutants to H2O2 and organic peroxides was significantly decreased. After introduction of the expression vector into the mutant and induction with 0.1% l-arabinose, resistance levels to oxidative stress in the rgsA-deleted mutants increased to the levels in the wild-type strain (Fig. 3).

Plate survival experiments of serially diluted Pseudomonas aeruginosa. Wild-type strain PAO1 (pJNT), rgsA deletion strain PAO1ΔrgsA (pJNT), and rgsA overexpression strain PAO1ΔrgsA (pJR1) cultured in LB medium alone or with hydrogen peroxide (H2O2), cumene hydroperoxide (CHP), or tert-butyl hydroperoxide (TBH), at the concentrations shown. The experiment was repeated three times
To study the effects of varying concentrations and durations of treatment with the oxidative stress-inducing substances on the growth of strains harboring the rgsA mutant, we employed the MTT assay and designed two low concentration (10 mM) and long duration (2 h, 5 h) and one high concentration (40 mM) and short duration (30 min) oxidative stress stimulation experiments using H2O2. With the increase in duration and concentration of H2O2, the number of surviving rgsA-deleted bacteria decreased compared with the number of surviving wild-type strain. Under short duration and high concentration stress conditions, the resistance of the overexpression strain was not recovered to the level of resistance of the wild-type strain (Fig. 4).

Viability of Pseudomonas aeruginosa strains exposed to hydrogen peroxide (H2O2). a Viability of the wild-type strain PAO1 (pJNT), rgsA deletion strain PAO1ΔrgsA (pJNT), and rgsA overexpression strain PAO1ΔrgsA (pJR1) after hydrogen peroxide (H2O2) treatment at different concentrations for different durations was estimated by measuring the absorbance. Results are presented as ratios compared to the survival of the untreated controls. Statistical significance was calculated using ANOVA (*P < 0.05; **P < 0.001). b The relative survival of the deletion and the overexpression strains compared to that of the wild-type strain is plotted vs. different time and concentrations of exposure to H2O2. Values are expressed as the mean ± SD; n = 3)
Sensitivity of P. aeruginosa Biofilm-type rgsA Mutants to Peroxide and Superoxide Stress
We used a flow apparatus to form biofilms after 3 d of culture. This was followed by the addition of different concentrations of H2O2 for in vitro simulation of oxidative stress. Thereafter, SYTO9/PI and fluorescence microscopy were performed. The number of dead bacteria in the rgsA-deleted mutants was more than that in the wild-type strain upon treatment with 20 mM H2O2, and the number was further increased upon treatment with 30 mM H2O2. However, the number of dead bacteria of the overexpression strain was similar to that of the wild-type strain upon treatment with the same H2O2 concentration, which is consistent with the oxidative stress responses observed in the planktonic state (Fig. 5).

Representative fluorescence microscopy images of Pseudomonas aeruginosa PAO1 (pJNT), rgsA deletion strain PAO1ΔrgsA (pJNT), and rgsA overexpression strain PAO1ΔrgsA (pJR1) after H2O2 treatment at different concentrations for 30 min in the biofilm state. Green represents live bacteria stained with Syto9, whereas yellow-orange represents dead bacteria stained with propidium iodide (scale bar = 100 μm) (Color figure online)
Discussion
In this study, we first determined the lengths of the rgsA transcripts and their transcription start sites. In a previous study, it was shown using northern blot analysis that rgsA is present as two transcripts of different lengths, approximately 90 and 300 bp, during the logarithmic phase but only one short transcript is present during the stationary phase [14]. All the RNA samples used in this study were obtained from P. aeruginosa in the pre-logarithmic phase (OD600 ≈ 0.5). Two transcripts of different lengths were detected in these RNA samples. It was difficult to obtain the full-length with 5′-RACE, and the PCR-recovered fragments had different start sites that varied by several dozens of bases but were indistinguishable. Therefore, sequences with different lengths were aligned with the P. aeruginosa rgsA gene sequence in the NCBI database, and the sequence providing the longest alignment was considered to have the start site at the 5′-end of the transcript. Based on the results of sequencing and sequence alignment, we concluded that there are two rgsA transcripts in P. aeruginosa with lengths of 90 and 300 bp, which is in agreement with the results of northern blot analysis [18]. The direction of transcription was the same in the two transcripts and common transcription stop sites may be present.
In this study, we focused on the role of rgsA in oxidative stress resistance. The real-time RT-PCR results showed that the expression levels of rgsA increased to varying degrees when cultures, at OD600 values of 0.5, were exposed to oxidative stress. rgsA deletion resulted in decreased resistance of P. aeruginosa in the planktonic and biofilm states to H2O2 and organic peroxides, and the resistance decreased with increasing concentrations of H2O2 and duration of the treatment. Decreased tolerance to the external environment may have influenced the growth rate and final concentration. The difference in growth rates between the rgsA-deleted mutant and wild-type strains was as high as 1.3 times of the growth rate in the logarithmic phase. The resistance to oxidative stress in the mutant strain could be partially restored to the resistance levels of the wild-type strain after introduction of the overexpression plasmid. Pei Lu demonstrated that during the exponential growth phase in rich medium, the expression level of RpoS was low and was repressed by rgsA, and a rgsA deletion modestly upregulated the RpoS protein level [19]. RpoS protects the bacteria against a variety of stressful conditions, including anaerobiosis, nitrogen and phosphate starvation, acid shock, osmotic stress, and oxidative stress [30]. However, in our study, the increased expression of the rpoS gene in the rgsA mutant was possibly unable to compensate for the decreased resistance of the rgsA deletion strain to oxidative stress. This suggests that rgsA may have more complex regulatory mechanisms. Pei Lu also showed that the overexpression of rgsA reduced the RpoS levels by 2.4-fold [19]. This may explain why the level of resistance to high concentrations of H2O2 in the mutant strain could not be restored to wild-type levels after introduction of the overexpression plasmid. Studies are underway in our laboratory to examine the altered protein expression of the rgsA deletion strain under oxidative stress.
Compared with other genes involved in the defense of P. aeruginosa against oxidative stress, including katB-ankB, ahpB, and ahpC-ahpF [31], the resistance of the rgsA mutant strain to oxidative stress was comparable to that of the wild-type strain, and the induction level of rgsA upon exposure to oxidative stress was quite modest. However, the regulation of sRNAs often contributes to the ability of bacteria to adapt to a changing environment quickly [32]. sRNAs are produced quickly, have the potential to propagate signals rapidly, regulate at the post-transcriptional level, and require less energy for synthesis than proteins [33,34,35] Furthermore, sRNAs have evolved diverse mechanisms [36] and affect multiple targets and/or proteins, which in turn act as regulators of other genes to improve the efficiency of regulation [37].
Based on the analysis of the DNA structure and biological function of rgsA, we understand that rgsA participates in various regulatory responses in bacteria, thereby affecting bacterial survival, metabolism, and pathogenicity. With the development of machine learning and various bioinformatics techniques, more sRNAs and their targets will be identified and validated, which is vital for a complete understanding of bacteria.
Conclusion
In this study, we show that deletion of the sRNA, rgsA, decreases the growth rate of P. aeruginosa. Furthermore, ΔrgsA P. aeruginosa exhibited a decreased ability to resist peroxide exposure, at different concentrations and for different durations, in both planktonic and biofilm growth modes compared with the wild-type strain. The results highlight the role of rgsA in the defense of P. aeruginosa against oxidative stress.
Data Availability
All data are fully available without restriction.
References
Imlay JA (2002) How oxygen damages microbes: oxygen tolerance and obligate anaerobiosis. Adv Microb Physiol 46:111–153. https://doi.org/10.1016/s0065-2911(02)46003-1
Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4(3):181–189. https://doi.org/10.1038/nri1312
Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ (2007) A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130(5):797–810. https://doi.org/10.1016/j.cell.2007.06.049
Lushchak VI (2001) Oxidative stress and mechanisms of protection against it in bacteria. Biochemistry 66(5):476–489. https://doi.org/10.1023/a:1010294415625
Cabiscol E, Tamarit J, Ros J (2000) Oxidative stress in bacteria and protein damage by reactive oxygen species. International microbiology : the official journal of the Spanish Society for Microbiology 3(1):3–8
Corona F, Martínez JL, Nikel PI (2019) The global regulator Crc orchestrates the metabolic robustness underlying oxidative stress resistance in Pseudomonas aeruginosa. Environ Microbiol 21:898–912. https://doi.org/10.1111/1462-2920.14471
Bjarnsholt T (2013) The role of bacterial biofilms in chronic infections. APMIS Suppl 121:1–58. https://doi.org/10.1111/apm.12099
Hentzer M, Eberl L, Givskov M (2005) Transcriptome analysis of Pseudomonas aeruginosa biofilm development: anaerobic respiration and iron limitation. Biofilms 2:37. https://doi.org/10.1017/S1479050505001699
Lazazzera BA (2005) Lessons from DNA microarray analysis: the gene expression profile of biofilms. Curr Opin Microbiol 8(2):222–227. https://doi.org/10.1016/j.mib.2005.02.015
Rybtke M, Hultqvist LD, Givskov M, Tolker-Nielsen T (2015) Pseudomonas aeruginosa biofilm infections: community structure, antimicrobial tolerance and immune response. J Mol Biol 427(23):3628–3645. https://doi.org/10.1016/j.jmb.2015.08.016
Hershberg R, Altuvia S, Margalit H (2003) A survey of small RNA-encoding genes in Escherichia coli. Nucleic Acids Res 31:1813–1820. https://doi.org/10.1093/nar/gkg297
Hoe CH, Raabe CA, Rozhdestvensky TS, Tang TH (2013) Bacterial sRNAs: regulation in stress. Int J Med Microbiol (IJMM) 303(5):217–229. https://doi.org/10.1016/j.ijmm.2013.04.002
Storz G, Vogel J, Wassarman KM (2011) Regulation by small RNAs in bacteria: expanding frontiers. Mol Cell 43(6):880–891. https://doi.org/10.1016/j.molcel.2011.08.022
Sonnleitner E, Haas D (2011) Small RNAs as regulators of primary and secondary metabolism in Pseudomonas species. Appl Microbiol Biotechnol 91:63–79. https://doi.org/10.1007/s00253-011-3332-1
Reinhart AA, Powell DA, Nguyen AT, O’Neill M, Djapgne L, Wilks A, Ernst RK, Oglesby-Sherrouse AG (2015) The prrF-encoded small regulatory RNAs are required for iron homeostasis and virulence of Pseudomonas aeruginosa. Infect Immun 83(3):863–875. https://doi.org/10.1128/IAI.02707-14
Janssen KH, Diaz MR, Golden M, Graham JW, Sanders W, Wolfgang MC, Yahr TL (2018) Functional analyses of the RsmY and RsmZ small noncoding regulatory RNAs in Pseudomonas aeruginosa. J Bacteriol 200(11):e00736-e817. https://doi.org/10.1128/JB.00736-17
Sonnleitner E, Gonzalez N, Sorger-Domenigg T et al (2011) The small RNA PhrS stimulates synthesis of the Pseudomonas aeruginosa quinolone signal. Mol Microbiol 80:868–885. https://doi.org/10.1111/j.1365-2958.2011.07620.x
González N, Heeb S, Valverde C et al (2008) Genome-wide search reveals a novel GacA-regulated small RNA in Pseudomonas species. BMC Genomics 9:167. https://doi.org/10.1186/1471-2164-9-167
Lu P, Wang Y, Hu Y, Chen S (2018) RgsA, an RpoS-dependent sRNA, negatively regulates rpoS expression in Pseudomonas aeruginosa. Microbiology 164:716–724. https://doi.org/10.1099/mic.0.000632
Lu P, Wang Y, Zhang Y, Hu Y, Thompson KM, Chen S (2016) RpoS-dependent sRNA RgsA regulates Fis and AcpP in Pseudomonas aeruginosa. Mol Microbiol 102:244–259. https://doi.org/10.1111/mmi.13458
Dötsch A, Eckweiler D, Schniederjans M et al (2012) The Pseudomonas aeruginosa transcriptome in planktonic cultures and static biofilms using RNA sequencing. PLoS ONE 7:e31092. https://doi.org/10.1371/journal.pone.0031092
Taylor RK, Manoil C, Mekalanos JJ (1989) Broad-host-range vectors for delivery of TnphoA: use in genetic analysis of secreted virulence determinants of Vibrio cholerae. J Bacteriol 171:1870–1878. https://doi.org/10.1128/jb.171.4.1870-1878.1989
Grudniak AM, Klecha B, Wolska KI (2018) Effects of null mutation of the heat-shock gene htpG on the production of virulence factors by Pseudomonas aeruginosa. Future Microbiol 13:69–80. https://doi.org/10.2217/fmb-2017-0111
Schweizer HD (1993) Small broad-host-range gentamycin resistance gene cassettes for site-specific insertion and deletion mutagenesis. Biotechniques 15:831–834. https://doi.org/10.1371/journal.pone.0031092
Köhler T, Curty LK, Barja F, van Delden C, Pechère JC (2002) Swarming of Pseudomonas aeruginosa is dependent on cell-to-cell signaling and requires flagella and pili. J Bacteriol 182:5990–5996. https://doi.org/10.1128/jb.182.21.5990-5996.2000
Hoang TT, Karkhoff-Schweizer RR, Kutchma AJ, Schweizer HP (1998) A broad-host-range Flp-FRT recombination system for site-specific excision of chromosomally-located DNA sequences: application for isolation of unmarked Pseudomonas aeruginosa mutants. Gene 212(1):77–86. https://doi.org/10.1016/s0378-1119(98)00130-9
Newman JR, Fuqua C (1999) Broad-host-range expression vectors that carry the L-arabinose-inducible Escherichia coli araBAD promoter and the araC regulator. Gene 227:197–203. https://doi.org/10.1016/s0378-1119(98)00601-5
Rio DC, Ares M Jr, Hannon GJ, Nilsen TW (2010) Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb Protoc. https://doi.org/10.1101/pdb.prot5439
Schmittgen TD, Livak KJ (2008) Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3(6):1101–1108. https://doi.org/10.1038/nprot.2008.73
Loewen PC, Hengge-Aronis R (1994) The role of the sigma factor sigma S (KatF) in bacterial global regulation. Annu Rev Microbiol 48:53–80. https://doi.org/10.1146/annurev.mi.48.100194.000413
Ochsner UA, Vasil ML, Alsabbagh E, Parvatiyar K, Hassett DJ (2000) Role of the Pseudomonas aeruginosa oxyR-recG operon in oxidative stress defense and DNA repair: OxyR-dependent regulation of katB-ankB, ahpB, and ahpC-ahpF. J Bacteriol 182(16):4533–4544. https://doi.org/10.1128/jb.182.16.4533-4544.2000
Chen J, Morita T, Gottesman S (2019) Regulation of transcription termination of small RNAs and by small RNAs: molecular mechanisms and biological functions. Front Cell Infect Microbiol 9:201. https://doi.org/10.3389/fcimb.2019.00201
Holmqvist E, Unoson C, Reimegård J, Wagner EG (2012) A mixed double negative feedback loop between the sRNA MicF and the global regulator Lrp. Mol Microbiol 84(3):414–427. https://doi.org/10.1111/j.1365-2958.2012.07994.x
Hussein R, Lim HN (2012) Direct comparison of small RNA and transcription factor signaling. Nucleic Acids Res 40(15):7269–7279. https://doi.org/10.1093/nar/gks439
Takahashi MK, Chappell J, Hayes CA, Sun ZZ, Kim J, Singhal V, Spring KJ, Al-Khabouri S, Fall CP, Noireaux V, Murray RM, Lucks JB (2015) Rapidly characterizing the fast dynamics of RNA genetic circuitry with cell-free transcription-translation (TX-TL) systems. ACS Synth Biol 4(5):503–515. https://doi.org/10.1021/sb400206c
Dutta T, Srivastava S (2018) Small RNA-mediated regulation in bacteria: a growing palette of diverse mechanisms. Gene 656:60–72. https://doi.org/10.1016/j.gene.2018.02.068
Altuvia S (2004) Regulatory small RNAs: the key to coordinating global regulatory circuits. J Bacteriol 186(20):6679–6680. https://doi.org/10.1128/JB.186.20.6679-6680.2004
Acknowledgements
We would like to thank Professor Bin Huang of the First Affiliated Hospital, Sun Yat-sen University for the P. aeruginosa PAO1, Professor Yihe Ge of Ludong University, Professor Kangmin Duan of Northwest University, Professor Ningyi Zhou of Wuhan Institute of Virology, Professor Jianqiang Lin of Shandong University, Professor Rui Zhou of Huazhong Agricultural University for the plasmids and strains.
Funding
This work was supported by the National Natural Science Foundation of China (NSFC), Reference H1912.
Author information
Authors and Affiliations
Contributions
The study concept and design, funding acquisition and the manuscript revision were performed by JZ, XS. The experiments performance, study implementation, data analysis and the manuscript draft were performed by SH and JZ. The study design, and interpretation of data were performed by XM, QH, LF, GZ, JH, YG, QX and XZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hou, S., Zhang, J., Ma, X. et al. Role of rgsA in Oxidative Stress Resistance in Pseudomonas aeruginosa. Curr Microbiol 78, 3133–3141 (2021). https://doi.org/10.1007/s00284-021-02580-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-021-02580-z