
Abstract
The first results of a study into the microbiomes of benthic invertebrates found in sites with seeps (containing methane, oil, or a combination of methane and mud) and an underwater low-temperature vent of Lake Baikal are presented. Microorganisms were detected in the intestine of an oligochaete from the cold methane seep using microscopy. Analysis of 16S rRNA gene libraries revealed that the highest diversity of microorganisms was found in the nematode microbiomes where the members of 11 phyla were identified. Some of the detected prokaryotes are methanogens, nitrifiers, and nitrogen fixators, while some are involved in the sulfur cycle. Methanotrophs were detected in the microbiomes of oligochaetes and chironomids. The microbiomes of nematodes, chironomids, and bathynellids are composed of members of the Bacteroidetes and Firmicutes phyla, which are related to the symbiotic bacteria found in insects and animals from other ecotopes. Microorganisms typically found in the water and sediments of Lake Baikal were also detected in the invertebrates microbiomes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hydrothermal vents and cold seeps are unique structures at the bottom of seas and oceans. Animals accumulate around these features to use the chemical compounds present there as energy sources [1]. Most animals from these ecotopes have symbionts that produce organic substances and enzymes [2], while the host provides them with access to the reduced elements (H2, H2S, CH4, O2, and CO2). Presumably, in some ecosystems, the role of symbiotic microorganisms is more significant than that of ones that live independently [3]. Bacteria that use the energy from oxidation of sulfur-containing inorganic compounds for autotrophic growth are most often involved in the symbiosis with marine animals and serve as a main food source [4]. There are also sulfur-oxidizing bacteria capable of fixing carbon and providing the host with amino acids and co-factors of protein biosynthesis [5]. Symbiosis with methanotrophic bacteria is less common. This symbiotic relationship has been detected in Mollusca, Porifera, and Annelida, and it enables animals from these phyla to live indirectly from methane [2]. There is little data on the symbiosis of benthic invertebrates with microorganisms that use nitrogen compounds as an energy source, or for growth. Microbes involved in nitrification, denitrification, and anaerobic ammonium oxidation (ANAMMOX) were reported in sponges and corals [6]. Subcuticular symbiotic bacteria co-evolving with Ophiactis balli ophiuroids and having a common nitrogen metabolism were also reported [7].
Less is known about the interactions of microorganisms in freshwater ecosystems than in marine ones. The microbiome affects the method of reproduction in snails [8]. It is known about the chemoautotroph-animal symbiosis between the freshwater cave and amphipod single phylotype of bacteria in the sulfur-oxidizing clade Thiothrix [9]. No symbiont methanotrophs were detected in animals from freshwater ecosystems, although some researchers suggest their presence in water bodies with high methane concentrations [2]. To date, only the presence of methanol-oxidizing endosymbionts has been proven in freshwater crustaceans [10].
In recent decades, numerous hydrate-bearing structures seeping mineralized gaseous fluids with various elemental compositions have been detected at the bottom of Lake Baikal. The sediments of such areas were found to have elevated concentrations of methane, sulfate ions, and trace elements [11], as well as a high density of benthic invertebrates and fish [12, 13]. The methane concentrations in the sediments of the freshwater seeps from Lake Baikal were an order of magnitude higher than those in marine sediments inhabited by methanotrophic symbionts. Values of δ13C show that the upper trophic levels of the benthic community found in the mud volcano (South Baikal) are rooted in methanotroph production [14]. According to the data on the ratio of stable carbon isotopes in tissues of benthic animals, biological productivity in these areas of Lake Baikal is maintained by both photosynthesis and chemosynthesis, as well as by methanotrophy [12]. The results of isotopic investigations of nitrogen in animal tissues revealed an active role of microbial symbionts involved in the cycling of this element [15]. The key role of nitrogen for energy production was also discussed in a metagenomics analysis of genomes of microbial communities in the near-bottom area of the bathypelagic zone of the lake [16]. The pathways through which methane and other compounds are included in food webs of benthic organisms in the deep zone of Lake Baikal remain poorly understood. Several studies indicated the presence of bacteria in benthic animals, and discussed their symbiotic, or symbiotrophic, relationships. Microscopy detected bacteria of the genus Cristispira (Spirochaetes) on the crystalline style of molluscs [17]. Bacteria that are likely ectosymbionts have been detected on the shell and egg masses of limpets in Frolikha Bay [13]. During the under-ice period, the water column had a high abundance of Baikal crustaceans, Epischura baicalensis, whose stomachs, according to the in situ hybridization, contained archaea [18]. Archaea can also be methane-producing symbionts, and a high concentration of methane was registered in the zone containing the Epischura baicalensis habitat.
In recent years, the presence of epibiotic consortia-containing protozoa and bacteria has been recorded on the surface of the bodies of Baikal-endemic amphipods [19]. The gut microflora of the animals is also not well understood, their metabolic processes and relationships in areas with elevated methane concentrations and other reduced compounds in Lake Baikal are not known.
Our studies aimed to (a) investigate, using microscopy and molecular methods, microorganisms associated with dominant benthic invertebrates that inhabit sediments of Lake Baikal. Specifically, the composition and localization of microorganisms at sites with that there is discharge of gaseous mineralized fluids were investigated and (b) assess possible symbioses in these ecotopes, taking into account geochemical conditions.
Materials and Methods
Study Objects
The sediment samples containing benthic organisms were taken during fieldworks onboard the RV “G.Yu. Vereshchagin” from 2014 to 2017 in different areas of Lake Baikal (Table S1, Fig. 1). The animals were sampled for analysis (Table S1). Then the animals were washed several times with sterile water immediately after sampling and examined under an MBS-10 binocular (Russia) and an Olympus CX-2 microscope (Japan) to identify epibionts (ciliates and microbial films) on the skin.

Sampling sites at Lake Baikal. Site 1: Posolsk Bank (52.03°N, 105.84°E); Site 2: Gorevoy Utes (53.30°N, 108.39°E); Site 3: Frolikha Vent (55.52°N, 109.78°E); Site 4: Kukui Canyon (52.59°N, 106.77°E); Site 5: Begul (52.73°N, 106.59°E)
Transmission Electron Microscopy (TEM)
The samples were fixed with 2.5% glutaraldehyde solution in 0.1 M cacodylate buffer added by 2 mM MgCl2. Fixation was carried out for two hours at 4 °C; postfixation—with 1% OsO4 in 0.1 M cacodylate buffer overnight. The samples were dehydrated in ethanol (30%, 50% 70%, 95%, and 100%) and acetone solutions. After that, it was poured into Araldite 502 Kit resin according to the manufacturer’s instructions. The polymerized blocks were cut using an Ultracut R ultramicrotome (Leica, Germany) with a thickness of 40–70 nm; the slices were stained with lead citrate solution. Microscopy of the preparations was carried out on a LEO 906E transmission microscope (Zeiss, Germany).
Light Microscopy
The samples were fixed with 4% paraformaldehyde in 0.1 M phosphate buffer overnight at 4 °C. Then, the samples were rinsed and dehydrated in ethanol (30%, 50%, 70%, 95%, and 100%). After that, the samples were placed in the solution of absolute ethanol and xylene (1:1) for one–two hours, then—in xylene for two–four hours. Next, the samples were placed in a xylene–paraffin solution (1:1) and left in an incubator overnight at 37 °C. After that, the samples were transferred to molten paraffin at 60 °C and left for three hours. Next, the samples were poured into paraffin blocks and cut 6–8 µm thick slices using a sledge microtome (KhZMA, Ukraine). To identify microorganisms in tissues, the slices underwent Gram staining. The slices were visualized on an Axio Imager M1light microscope (Zeiss, Germany).
Extraction of Total DNA and Amplification
Total DNA was extracted by a modified enzymatic lysis method followed by phenol–chloroform extraction. Fragments of the 16S rRNA gene from the total DNA of benthic animals were amplified using standard bacterial 27F:5′-AGAGTTTGATCCTGGCTCAG, 1350R:5′-GACGGGCGGTGTGTACAAG, and archaeal, 21F:5′-CCCGGTTGATCCYGCCRG, 958R:5′-YCCGGCGTTGAMTCCAATT primers [20, 21]. Polymerase chain reaction (PCR) was carried out using AmpliSens kits (Central Research Institute of Epidemiology of the Federal Service on Customers’ Rights Protection and Human Well-being Surveillance, Russia) according to the manufacturer’s instructions. The resulting PCR products were ligated and cloned using a GeneJET™ PCR Cloning Kit (Fermentas, Lithuania) according to the manufacturer’s instructions. Nucleotide sequences of the insertions were determined for 271 independent clones.
Denaturing Gradient Gel Electrophoresis (DGGE)
Fragments of the 16S rRNA gene from the total DNA of benthic animals were amplified using standard bacterial primers, 40-bp GCclamp-341F:5′-CCTACGGGAGGCAGCAG and 907R:5′-CCGTCAATTCCTTTRAGTTT [22]. PCR amplification products were applied directly to 8% (in volume) polyacrylamide gel with a 40–70% concentration gradient of acrylamide in 0.5xTAE buffer solution according to the instructions of the manufacturer of the DCode device (Bio-Rad, USA). Electrophoresis was carried out at a constant voltage of 70 V and temperature of 60 °C during 16–17 h. After staining with ethidium bromide, individual bands were cut out and eluted in 20 µl of sterile water. Using eluted DNA as a matrix, PCR was carried out with the same primers without GC-end.
Sequencing
The Sanger sequencing was carried out using the BigDye Terminator Kit v.3.1 reagent on an ABI 3130XL Genetic Analyzer (Applied Biosystems, USA) in SB RAS Genomics Core Facility (Institute of Chemical Biology and Fundamental Medicine Siberian Branch of the Russian Academy of Sciences, Novosibirsk).
Phylogenetic Analysis
The resulting sequences were compared with the sequences from the international NCBI database using the BLASTN program (http://www.ncbi.nlm.nih.gov/blast). Identical sequences were grouped into operational taxonomic units (OTUs). The presence of chimeras was determined through the sequence analysis using the PINTAIL program (http://www.cardiff.ac.uk/biosi/research/biosoft). The structures were analyzed using the ClustalW1 program (http://www.ebi.ac.uk/clustalw). Comparison of sequences and construction of phylogenetic trees were carried out using the MEGA6 software and the neighbor-joining algorithm. Statistical significance of branching was evaluated by bootstrapping.
The sequences were deposited in GenBank under accession Nos MN227537-MN227539, MN235838-MN235841, MN240819-MN240833, MN241248-MN241252, MN240877-MN240880, MN448346-MN448358, MN647049-MN647061, MN822696-MN822698, MN453528, MN453529, and MN865594.
Results
Microscopy
Examination of the studied animals under the light microscope did not reveal epibiotic consortia of protozoa and bacteria on the skin of all animal samples. Following staining with methyl violet and fuchsin, gram-positive microorganisms were only detected in the intestinal region of oligochaetes from the site with methane seep (Fig. 2). The cells stained a dark blue color had the form of coccobacilli with dimensions of ~ 2.5 × 1.8 µm (n = 10). Most studied oligochaetes had empty intestines, which is most likely due to the effect of stress after moving the animals from great depths (330–1169 m).

Slice of an oligochaete (Site 1). Light microscopy. a Rectangle marks a fragment of the accumulation of bacterial cells in the tissue b a magnified fragment indicated in the photo, arrows show the accumulations of Gram-positive microorganisms
Analysis of ultrathin slices (18 samples) using TEM confirmed the presence of bacterial cells in tissues of oligochaetes. Figure 3a shows the cells with dimensions of 0.62 µm (n = 8). There was a vacuolization of cells, which was especially evident in muscle tissues. It is most likely caused by the stresses that animals experience when they ascend from great depths to the water surface.

a Ultrathin slice of an oligochaete (Site 1). Arrows show cells of putative microorganisms. b Ultrathin slice of a nematode (Site 1). The arrow shows a putative microorganism in cuticle tissues
Analysis of ultrathin slices of nematodes indicated the presence of single cells located in cuticle tissues (Fig. 3b), whose dimensions did not exceed 200 nm.
Molecular Biological Analysis
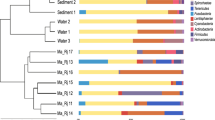
Analysis of 16S rRNA gene libraries resulted in obtaining the sequences of 11 bacterial and 2 archaeal phyla (Table S1, Fig. 4A). All studied animals had members of the class Gammaproteobacteria (Fig. 5). The nematode microbiomes contained the highest number of phyla (11), and bathynellids contained the lowest. Analysis of the sequences indicated one common OTU in the microbiomes of nematodes and oligochaetes (Sphingomonas sp.) as well as one in nematodes and chironomids (Dysgomonas sp.). In the oligochaete microbiomes from four sites, common members were at the phylum level (Proteobacteria), whereas in the nematode microbiomes, common members were at the class (Alphaproteobacteria, Deinococci, Gammaproteobacteria) and genus (Acinetobacter sp.) levels.


a Neighbor-joining phylogenetic analysis of 16S rRNA gene sequences of bacteria and archaea, associated with oligohaetes (circle, number inside means site), chironomids (triangle), batinellids (black circle), and their closest relatives. b Neighbor-joining phylogenetic analysis of 16S rRNA gene sequences of bacteria and archaea, associated with nematodes (square, number inside means site) and their closest relatives

Microorganisms separation into symbionts and food source, according to their occurrence in the sediments and water column of the lake, as well as by analysis of the 16SrRNA gene homologs
Nematodes are among the most common benthic animals in Lake Baikal. We analyzed nematode microbiomes from two sites (1 and 2). Bacteria of the genus Acinetobacter (Gammaproteobacteria) were commonly represented in the nematode microbiomes. These bacteria comprised 22% of the site 1 clone library, which may be due to the dominance of this genus in the sediments of this area (16%) [23]. Single sequences were identified in the site 2 clone library, where members of Acinetobacter were also recorded. We did not identify members of this genus in the microbiomes of other studied animals.
We detected members of the phylum Deinococcus-Thermus in nematode microbiomes from two sites. In Lake Baikal, this phylum was previously detected in neuston (LN736018), sediments adjacent to gas hydrates [23], near-bottom deep layers, where an algicidal effect on the diatom S. acus took place [24]. In sediments and nematodes from both sites, the genus Deinococcus represented this phylum, and in the nematode microbiome of Site 1, we detected the sequence belonging to the genus Thermus.
The third common representative of the nematode microbiomes was bacteria of the genus Sphingomonas (Alphaproteobacteria). The members of this genus are widespread in sediments and water of the lake, and are involved in biodegradation of complex organic compounds. However, the species Sphingomonas melonis represents the closest homologs for the Site 1 nematodes, which were isolated from the soils (MT043729, MN177231) and water column affected by the Gulf oil (MT515445). The species Sphingomonas echinoides represent the closest homologs for the Site 2 nematodes, which were detected in bacterial associates of scale insects (MK462263) and silkworm litter (LC484826), as well as in deep-sea sediments (MH725538).
In the Site 1 nematode microbiomes, we detected bacterial sequences of the phyla Nitrospira and Acidobacteria, which are present in the sediments of many Baikal sites [23, 25]. The former ones are nitrifiers, and the latter ones are decomposers of biopolymers. The microbiomes of these animals contained members of the species Rhodopseudomonas (Alphaproteobacteria), which are phototrophic nitrogen fixators; however, their presence was not observed in the lake sediments. Representatives of this genus were found in the posterior intestinal tract of the polychaete Neanthes glandicincta (FJ618865), though its contribution is not known. Moreover, the Site 1 nematode microbiomes had the members of the genera Variovorax and Pelomonas (Betaproteobacteria), which were not previously detected in the sediments of this site. In Lake Baikal, bacteria of the genus Variovorax were identified in the communities associated with the colorless sulfur bacteria (HQ400611). Their physiological and biochemical properties suggest involvement in the sulfur and nitrogen cycle.
The bacterial sequences belonging to the phyla Bacteroidetes (Dysgomonas sp., 24%) and Actinobacteria (18%), which are involved in the decomposition of organic substances, dominated the clone library of the Site 2 nematodes. Bacteria of the genus Dysgomonas were not previously identified in the ecosystem of the lake and the closest homolog is the alginate-degrading bacteria, Dysgonomonas alginatilytica sp. nov. (NR137388). Actinobacteria are widespread, and in the sediments of this site, they comprised a third of the entire microbial community [25].
The Site 2 nematode microbiomes showed the sequences typical of the representatives of oligotrophic ecosystems: bacteria of the species Caulobacter (Alphaproteobacteria) and Limnohabitans (Betaproteobacteria). Prostecobacteria were often detected in the water column and sediments of the lake [25, 26]. The closest homolog for these bacteria was isolated from epidermal mucus layers of stingrays (KP713643). The presence of bacteria of the genus Limnohabitans was recorded in neuston and photic layers of Lake Baikal [16, 27, 28]. Bacteria of this genus have high rates of growth and assimilation of organic matter, and, being a food source for protists, they are an important link in the carbon flow to the upper trophic layers [29]. Furthermore, the microbiomes of these nematodes had bacteria of the phylum Firmicutes (15%), as well as cyanobacteria of the genus Synechococcus. While the latter dominate in Lake Baikal in summer and are found in all seasons in the photic zone [30], they formed only a minor proportion of the bacteria in the sediments of this site. Firmicutes of the genera Clostridium sp. and Finegoldia sp. were also detected in the sediments of this site. The clostridial sequences from nematodes were close to the bacterial sequences from the digestive tract of chironomids (LN828873). Site 2 nematode microbiomes also contained hydrogenotrophic methanogenic archaea of the phylum Euryarchaeota (class Methanomicrobia), which were previously detected in the sediments of this site (SRP052288). Oligochaetes are the second group with a high population density at the discharge sites. The taxonomic composition differed between the oligochaete microbiomes from four sites. In the Site 1 oligochaete microbiome, we identified members of Alphaproteobacteria (Methylocystaceae). The closest homologs are involved in the methane oxidation (AY203783, AB930623). In the phylogenetic tree, these sequences form a separate cluster (Fig. 4) within Alphaproteobacteria as they show a low similarity with cultured homologs (91%), and high similarity with uncultured ones (97–99%). Representatives of the family Methylocystaceae were often detected in the communities of the bottom sediments from different sites of Lake Baikal [25]. The oligochaete microbiome of Site 1 also had Planctomycetes, whose closest homologs are involved in nitrogen metabolism, and Thaumarchaeota that are involved in aerobic ammonium oxidation. In the deep zone of the water column, the members of these taxa were up to 10 and 20% of the total 16S rRNA reads, respectively (according to metagenome-assembled genomes) [16], and in the surface sedimentary layer of this site, formed 2 and 40%, respectively [23]. Some Thaumarchaea contain the genes to synthesize methylphosphonate, a precursor of methane production in the ocean [31]. In general, the closest Thaumarchaeota homologs from sediments and oligochaetes coincide.
In the total DNA of the Site 2 oligochaete, there were sequences of Gammaproteobacteria identical to the sequences of bacteria from the gastrointestinal tract of the Neanthes glandicinta polychaeta (FJ618865, FJ618883). Moreover, in the oligochaete microbiome, we identified the sequences showing similarity (97%) with colorless sulfur bacteria of the genus Thiothrix (Gammaproteobacteria). The sequences of the 16S rRNA gene of the previously identified Thiothrix members differ from each other in the ostracod microbiome from the lake and hot spring (DQ780007, MN602536 and MN602537), which indicate their belonging to different species. Previously, another genus of colorless sulfur bacteria, Thioploca, was recorded at this site [32].
In the Site 3 oligochaete microbiome, there were members of the family Comamonadaceae (Betaproteobacteria) and the genus Thioploca (Gammaproteobacteria) and Actinobacteria. During the dives of the deep-sea manned submersibles at this site, bacterial mats were described [12]. In the microbial community of the sediments at this site, 44% of the sequences belonged to the phylum Actinobacteria, while Betaproteobacteria comprised only 2% and had members of other orders (Rhodocyclaceae and Methylophilaceae).
In the total DNA of an oligochaete (Site 5), we identified the sequences of the bacteria of the genus Sphingomonas, whose closest homologs were recorded in the gender-specific bacterial composition of black flies (JF733344). Bacteria of the genus Sphingomonas are found in ecotopes with low nutrient concentrations; they can biodegrade toxic substances and pollutants. In the sediments of Lake Baikal, this genus was not previously recorded, although bacteria of the family Sphingomonadaceae were detected. The microbiome of the same oligochaete showed thermophilic bacteria of the genus Thermaerobacter (Firmicutes), whose the presence was previously recorded in the sediments of the Posolsk Bank methane seep (KY492688).
In the microbial community of the Site 3 chironomids, we revealed the sequences of Gamma- and Deltaproteobacteria that differ in structure from those in microbiomes of other animals. Thus, the order Methylococcales represented the Gammaproteobacteria class and was present both whose the presence was recorded both in the bottom sediments and water column of Lake Baikal [25]. The closest homologs of methanotrophs from chironomids were found in the ecotopes rich in Fe and/or Mn (GU572365, AB722196). The Methylococcales episymbionts expressed the soxBXY and sqr genes implying that they can also oxidize reduced sulfur compounds [33].
We observed members of the genus Desulfovibrio (Deltaproteobacteria) only in the chironomid microbiome, but their presence was also indicated by immunofluorescence in the sediments of Northern Baikal [12]. A quarter of the sequences from the chironomid clone library belongs to the phylum Bacteroidetes. The closest homologs were isolated from the digestive tract of medicinal leeches (EU616637) and chironomids (mineral Lake Chany, Novosibirsk Region, LN828857). The closest cultured homolog (97%), the Mucinivorans hirudinis anaerobe (KM272750), is a mucin-degrading bacterium from the digestive tract of medicinal leeches. The cultured members of the family Rikenellaceae were isolated from the digestive tract of animal feces and are anaerobes and mesophiles that function to split carbohydrates and proteins. It was suggested [34] that the members of this family are specialized in symbiosis and can colonize the digestive organs in a wide spectrum of animals, from invertebrates to mammals, including humans. Furthermore, a group of bacterial sequences belonging to the genus Dysgonomonas (Bacteroidetes) was identified in the chironomid microbiome, different species of which were isolated from the digestive tracts of termites (LC021528). In the chironomid microbiome, we identified three OTUs of bacteria of the phylum Firmicutes, which differed from those identified in nematodes. They all belonged to the class Clostridia and are mainly obligate spore-forming anaerobes. The closest homologs were isolated from the posterior intestines of crabs and beetle larvae (HG792226, FJ374230), and the digestive tract of insects (JN653032) and fish (DQ816804). The members of this phylum were consistently detected in small amounts (less than 3.5%) in the water and sediments at different sites of Lake Baikal [25, 28].
Lake Baikal is the only open water body inhabited by bathynellids, whose habitat extends from the wave splash zone to the maximum depths. Their microbiomes (Site 4) indicated the sequences of bacteria of the genus Variovorax (Betaproteobacteria), whose closest homologs are endophytic bacteria of the halophyte (KF844058). Moreover, the bathynellids microbiome contained members of the genus Bradyrhizobium (Alphaproteobacteria), whose closest homolog (LC218372) was isolated from the soil of the rice rhizosphere during cultivation of lanthanum-dependent methylotrophs. These microorganisms have cosmopolitan distribution and are strict anaerobes. In the bathynellids microbiome, we also identified the members of the families Sphingobacteriaceae and Amoebophilaceae (Bacteroidetes). The closest homologs for the sequences of the former family were recorded in natural samples with a wide temperature range, for example, in the soils of greenhouses (NR_044568) and the Antarctic (KY405905). The members of the latter family belonged to the genus Cardinium (MH057615). These are cytoplasmatic, symbiotic bacteria that are widespread in terrestrial arthropods and regulate the development and reproduction of their hosts [35]. They have not been previously found in the sediments and water of the lake.
Discussion
In the studied sediments of Lake Baikal, which are confined to the discharge zones of mineralized fluids saturated with hydrocarbon gases, we recorded a wide spectrum of benthic animals with different densities [36, 37]. In the ocean, the animal biomass in oases of life is 2–5 orders of magnitude higher than the biomass at reference sites, due to nutrition availability [38]. There is the same pattern in Lake Baikal, where a greater diversity of invertebrates is seen in the sediments where there is discharge of gas-containing mineralized fluids of different compositions. The largest number of nematode species was recorded near the Gorevoy Utes oil and methane seep, at sites adjacent to oil seepages [32]. For the oligochaete species, their maximal densities were observed in the underwater hot spring of the Frolikha Bay [39].
Methane is involved in food webs of marine and freshwater ecosystems, both through symbionts [40, 41] and through consumption of biomass by methane-oxidizing bacteria. As observed in laboratory experiments, chironomid leeches and the Daphnia planktonic crustaceans are particularly important for these process [42, 43]. According to [37], the proportion of assimilated methane carbon in some animals reached 89%, which indicates the consumption of methanotrophic bacteria that are highly abundant in methane seep. In Lake Baikal, abnormally low values of δ13C (−52 ± −62‰) are observed in the tissues of different groups of benthic animals in discharge sites indicating the involvement of methane carbon in food chains [14, 33]. Light δ13C signatures were recorded for chironomides (−52 ± −62‰), oligochaetes (−62.5 ± 4.7‰), and flatworms (−63.9 ± 1.0‰) and were very variable in the tissues of sponges (−65 ± −37.3‰) [15, 36]. Lighter δ13C signatures (up to −75‰) were found in tissues of gastropoda, amphipoda, and chironomidae from the Frolikha vent (Site 3) [36]. High concentrations and rates of methane oxidation, as well as a high density of chironomids and other animals, were repeatedly recorded at this site [25]. Here, type I methanotrophs were detected in the Site 3 chironomid microbiome. At Site 1, where oligochaetes were dominant among all benthic animals, the sequences of type II methanotrophs were identified. Oligochaetes pass soil, which is from 4 to 6 times heavier than the own weight, through their digestive system, thus, contributing to oxygen penetration into sediments, assimilation of microflora, and intensive processes of mineralization of organic matter in the lake. Previously, there was almost no mention about the symbiosis of animals with type II methanotrophs, which can be explained by their lower energy efficiency compared to type I methanotrophs [44]. Furthermore, type II methanotrophs develop in ecotopes with higher methane concentrations and lower oxygen and nitrogen concentrations than type I methanotrophs [45]. There is only one known case of symbiotic type II methanotrophs inhabiting sphagnum moss cells [46], whose biomass consists of 15% of methane carbon, obtained from the activity of methane-oxidizing bacteria.
We assume that the main route of methane carbon introduction into Lake Baikal is through the consumption of methane-oxidizing bacteria as food. By forming areas of high biomass in microbial mats at sites with methane discharge, methanotrophs and other bacteria can ensure the coexistence of different animal species in the bathypelagic zone of Lake Baikal [47].
It is worth noting that microorganisms involved in the sulfur cycle were present in the microbiomes of the studied animals. As a rule, elevated concentrations of sulfate ions are observed only at the sites with the discharge of mineralized gas-containing fluids [11]. Outside these sites, in the sediments and water column of Lake Baikal, concentrations of sulfate ions are less than 5–10 mg/l, and the concentration of hydrogen sulfide is below the sensitivity threshold of the methods applied [48]. In the near-bottom water of Lake Baikal, the SOX oxidation pathway was mainly observed [16], whereas the presence of the SOX genes was typical of symbionts of the animals from vents [49].
In marine ecosystems, nematodes form symbiotic relationships with sulfur-oxidizing bacteria located on their cuticle. Although we did not identify epibiotic consortia on the skin of the studied animals, the oligochaete microbiomes were composed of chemo-myxotrophic colorless sulfur bacteria of the genera Thiothrix and Thioploca (Site 2 and 3, respectively), as well as chemoautotrophic purple sulfur bacteria of the family Chromatiaceae (Site 3). Bacteria of the genus Thiothrix have a symbiotic relationship with amphipods [9] and Kiwa hirsuta deep-sea crabs [4] and serve as their food source. It is likely that the colorless sulfur bacteria of the genera Thioploca and Thiothrix protect animals from the toxic effects of hydrogen sulfide. The Thiotrichales episymbionts are able to fix carbon in the absence of electron donors, using intracellular sulfur globules [33]. It cannot be excluded that colorless sulfur bacteria are also food for the Baikal oligochaetes, and this may explain their high density at the site with microbial mats [12, 36].
The microbiomes of all species of the studied animals also contain of microorganisms involved in the nitrogen cycle. As shown Baikalian Thioploca is characterized by a light δ15N signature, because of its active metabolism of mineral nitrogen during nitrate respiration [50]. The presence of various bacteria, including methanotrophs, in the sheaths of Thioploca and a light δ13C signature are evidence of methane carbon consumption. In the oligochaete, bacteria involved in the nitrogen cycle were firstly bacteria of the phylum Planctomycetes were firstly bacteria of the phylum Planctomycetes, some members of which can carry out anaerobic ammonium oxidation to nitrogen and can degrade hydrocarbons produced by phytoplankton [51]. Second, chemolithotrophic archaea of the phylum Thaumarchaeota are also involved in nitrogen cycling and can carry out aerobic ammonium oxidation to nitrogen. Thaumarchaea are ubiquitous, found in the water columns of oligotrophic lakes [31] as well as in symbiotic relationships with sponges, corals and ascidians, where they utilize ammonium and provide a detoxification mechanism for tissues [52]. Also, they produce cobalamin, the precursor to vitamin B12, for sharing with other organisms [31]. In addition, archaea can be the basal producers in the food chain due to their ability to scavenge dead cells [53]. Thaumarchaea have been detected within the food vacuoles of ciliates and dinoflagellates [31].
Figure 5 shows the diversity of microorganisms associated with various taxa of Baikal animals, taking into account possible symbioses (the presence of symbionts among the closest homologs, as well as their presence or absence in the sediments and water). A comparative analysis of the taxonomic composition of microorganisms indicates a wide spectrum of microorganisms associated with animals. The nematode microbiomes are the most diverse. It is also evident that the studied animals do not completely depend on the activity of symbionts. The bulk of microorganisms detected in the microbiomes of the animals were found in the communities of the bottom sediments and water column of Lake Baikal. In the animals of the same species from different sites, we recorded different numbers of taxa and accompanying microorganisms. The members of the phyla Bacteroidetes and Firmicutes identified in the microbiomes of nematodes, chironomids, and bathynellids were similar to the sequences of the symbiotic bacteria identified in various insects and animals.
Conclusion
We think that microorganisms identified in the animal microbiomes and studies ecotype can be considered as food sources, rather than as symbionts. In general, the microbiomes of studied benthic invertebrates from Lake Baikal consist of microorganisms that are capable of independent existence in the environment and can contribute to the splitting of complex compounds.
References
Dick GJ (2019) The microbiomes of deep-sea hydrothermal vents: distributed globally, shaped locally. Nat Rev Microbiol 17:271–283. https://doi.org/10.1038/s41579-019-0160-2
DeChaine EG, Cavanaugh CM (2005) Symbioses of methanotrophs and deep-sea mussels (Mytilidae: Bathymodiolinae). In: Overmann J (ed) Molecular basis of symbiosis progress in molecular and subcellular biology, vol 4. Springer, Berlin, pp 227–249
Beinart RA (2019) The significance of microbial symbionts in ecosystem processes. mSystems. https://doi.org/10.1128/mSystems.00127-19
Goffredi SK, Jones WJ, Erhlich H, Springer A, Vrijenhoek RC (2008) Epibiotic bacteria associated with the recently discovered Yeti crab, Kiwa hirsute. Environ Microbiol 10:2623–2634. https://doi.org/10.1111/j.1462-2920.2008.01684.x
Ponnudurai R, Kleiner M, Sayavedra L, Petersen JM, Moche M, Otto A, Becher D, Takeuchi T, Satoh N, Dubilier N, Schweder T, Markert S (2017) Metabolic and physiological interdependencies in the Bathymodiolus azoricus symbiosis. ISME J 11:463–477. https://doi.org/10.1038/ismej.2016.124
Fiore CL, Jarett JK, Olson ND, Lesser MP (2010) Nitrogen fixation and nitrogen transformations in marine symbioses. Trends Microbiol 18:455–463. https://doi.org/10.1016/j.tim.2010.07.001
Burnett WJ, McKenzie JD (1997) Subcuticular bacteria from the brittle star Ophiactis balli (Echinodermata: Ophiuroidea) represent a new lineage of extracellular marine symbionts in the alpha subdivision of the class Proteobacteria. Appl Environ Microbiol 63:1721–1724. https://doi.org/10.1128/AEM.63.5.1721-1724.1997
Takacs-Vesbach C, King K, Van Horn D, Larkin K, Neiman M (2016) Distinct bacterial microbiomes in sexual and asexual Potamopyrgus antipodarum, a New Zealand freshwater Snail. PLoS ONE 11(8):e0161050. https://doi.org/10.1371/journal.pone.0161050
Flot JF, Wörheide G, Dattagupta S (2010) Unsuspected diversity of Niphargus amphipods in the chemoautotrophic cave ecosystem of Frasassi, central Italy. BMC Evolut Biol 10:171. https://doi.org/10.1186/1471-2148-10-171
Mioduchowska M, Czyż MJ, Gołdyn B, Kilikowska A, Namiotko T, Pinceel T, Laciak M, Sell J (2018) Detection of bacterial endosymbionts in freshwater crustaceans: the applicability of non-degenerate primers to amplify the bacterial 16S rRNA gene. PeerJ 6:e6039. https://doi.org/10.7717/peerj.6039
Granina LZ (2008) Early diagenesis of bottom sediments in Lake Baikal. Academic Publishing House Geo, Novosibirsk
Namsaraev BB, Dulov LE, Dubinina GA, Zemskaya TI, Granina LZ, Karabanov EV (1994) Participation of bacteria in processes of synthesis and destruction of organic matter in microbial mats of the Lake Baikal. Microbiology 63:344–351
Sitnikova TY, Shirokaya AA (2013) New data on deep-water Baikal limpets found in hydrothermal vents and oil-seeps. Archiv fuer Molluskenkd 142:257–278. https://doi.org/10.1127/arch.moll/1869-0963/142/257-278
Klerkx J, Zemskaya TI, Matveeva TV, Khlystov OM, Namsaraev BB, Dagurova OP, Golobokova LP, Vorob’yova SS, Pogodaeva TP, Granin NG, Kalmichkov GV, Ponomarchuk VA, Shodgi H, Mazurenko LL, Kaulio VV, Solov’yov VA, Grachev MA (2003) Methane hydrates in deep bottom sediments of Lake Baikal. Dokl Earth Sci 393:822–826
Sitnikova T, Kiyashko S, Bukshuk N, Zemskaya T, Khlystov O, Moore MV (2016) Stable isotope signatures and distribution of deepwater sponges in Lake Baikal. Hydrobiologia 773:11–22. https://doi.org/10.1007/s10750-016-2674-1
Cabello-Yeves PJ, Zemskaya TI, Zakharenko AS, Sakirko MV, Ivanov VG, Ghai R, Rodriguez-Valera F (2020) Microbiome of the deep Lake Baikal, a unique oxic bathypelagic habitat. Limnol Oceanogr 65:1471–1488. https://doi.org/10.1002/lno.11401
Tulupova YuR, Parfenova VV, Sitnikova TYa, Sorokovnikova EG, Khanaev IB (2012) First report on bacteria of the family Spirochaetaceae from digestive tract of endemic gastropods from Lake Baikal. Microbiology 81:500–507. https://doi.org/10.1134/S0026261712030150
Maximenko SYu, Zemskaya TI, Naumova YuYu, Melnik NG (2008) Analysis of the microbial community of the stomach of the Baikal endemic Epischura baicalensis using the method of fluorescence in situ hybridization. Hydrobiol J 44:78–82. https://doi.org/10.1615/HydrobJ.v45.i1.60
Khalzov IA, MekhanikovaSitnikova IVTYa (2018) First data on ectosymbiotic consortia of infusoria and prokaryotes associated with amphipods inhabiting the Frolikha underwater hydrothermal vent, lake Baikal. Zool zhurnal 97:1525–1530. https://doi.org/10.1134/S0044513418120073
Denisova LYa, Bel’kova NL, Tulokhonov II, Zaichikov EF (1999) Bacterial diversity at various depth in the southern part of lake Baikal as revealed by 16S rDNA sequencing. Microbiology 68:475–483
Hallam SJ, Girguis PR, Preston CM, Richardson PM, DeLong EF (2003) Identification of methyl coenzyme M reductase A (mcrA) genes associated with methane-oxidizing archaea. Appl Environ Microbiol 69:543–549. https://doi.org/10.1128/AEM.69.9.5483-5491.2003
Muyzer G, Teske A, Wirsen CO (1995) Phylogenetic relationships of Thiomicrospira species and their identification in deep-sea hydrothermal vent samples by denaturing gradient gel electrophoresis of 16S rDNA fragments. Arch Microbiol 164:165–172
Chernitsyna SM, Mamaeva EV, Lomakina AV, Pogodaeva TV, Galach’yants YuP, Bukin SV, Pimenov NV, Khlystov OM, Zemskaya TI (2016) Phylogenetic diversity of microbial communities of the Posolsk Bank bottom sediments, Lake Baikal. Microbiology 85:652–662. https://doi.org/10.1134/S0026261716060060
Zakharova YuR, Kurilkina MI, Likhoshvay AV, Shishlyannikov SM, Kalyuzhnaya OV, Petrova DP, Likhoshway EV (2013) Effect of bacteria from the bottom water layer of Lake Baikal on degradation of diatoms. Paleontol J 47(9):1030–1034. https://doi.org/10.1134/S0031030113090256
Zemskaya TI, Lomakina AV, Shubenkova OV, Pogodaeva TV, Morozov IV, Chernitsina SM, Sitnikova TY, Khlystov OM, Egorov AV (2015) Jelly-like microbial mats over subsurface fields of gas hydrates at the St. Petersburg methane seep (Central Baikal). Geomicrobiol J 32:89–100. https://doi.org/10.1080/01490451.2014.910572
Lapteva IA (1987) Environmental features of the distribution of bacteria Caulobacter genus in fresh water basins. Microbiology 56:677–684
Galach’yants AD, Bel’kova NL, Sukhanova EV, Galach’yants YP, Morozova AA, Parfenova VV (2017) Taxonomic composition of Lake Baikal bacterioneuston communities. Microbiology 86:241–249. https://doi.org/10.1134/S0026261717020084
Cabello-Yeves PJ, Zemskaya TI, Rosselli R, Coutinho FH, Zakharenko AS, Blinov VV, Rodriguez-Valera F (2018) Genomes of novel microbial lineages assembled from the sub-ice waters of Lake Baikal. Appl Environ Microbiol 84:e02132-e2217. https://doi.org/10.1128/AEM.02132-17
Kasalický V, Jezbera J, Hahn MW, Šimek K (2013) The diversity of the Limnohabitans genus, an important group of freshwater bacterioplankton, by characterization of 35 isolated strains. PLoS ONE 8:e58209. https://doi.org/10.1371/journal.pone.0058209
Popovskaya GI, Belykh OI (2003) Stages of studying the autotrophic picoplankton of Lake Baikal. Hydrobiol J 39:12–24
Santoro AE, Richter RA, Dupont CL (2019) Planktonic marine Archaea. Annu Rev Mar Sci 11:131–158. https://doi.org/10.1146/annurev-marine-121916-063141
Naumova TV, Sitnikova TY, Gagarin VG (2012) The species composition and distribution of free-living Nematodes (Nematoda) in an area of natural oil and gas seeps in Lake Baikal. Inland Water Biol 5:161–168. https://doi.org/10.1134/S1995082912020113
Motoki K, Watsuji T, Takaki Y, Takai K, Iwasaki W (2020) Metatranscriptomics by in situ RNA stabilization directly and comprehensively revealed episymbiotic microbial communities of deep-sea squat lobsters. mSystems 5:e00551-e620. https://doi.org/10.1128/mSystems.00551-20
Worthen PL, Gode CJ, Graf J (2006) Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis. AEM 72:4775–4781. https://doi.org/10.1128/AEM.00356-06
Gotoh T, Noda H, Ito S (2007) Cardinium symbionts cause cytoplasmic incompatibility in spider mites. Heredity 98:13–20. https://doi.org/10.1038/sj.hdy.6800881
Zemskaya TI, Sitnikova TY, Kiyashko SI, Kalmychkov GV, Pogodaeva TV, Mekhanikova IV, Naumova TV, Shubenkova OV, Chernitsina SM, Kotsar OV, Chernyaev ES, Khlystov OM (2012) Faunal communities at sites of gas- and oil-bearing fluids in Lake Baikal. Geo-Mar Lett 32:437–451. https://doi.org/10.1007/s00367-012-0297-8
Sitnikova TYa, Mekhanikova IV, Sideleva VG, Kiyashko SI, Naumova TV, Zemskaya TI, Khlystov OM (2017) Trophic relationships between macroinvertebrates and fish in St. Petersburg methane seep community in abyssal zone of Lake Baikal. Contemp Probl Ecol 10:147–156. https://doi.org/10.1134/S1995425517020123
Vanreusel A, Andersen A, Boetius A, Connelly D, Cunha M, Decker C, Hilario A, Kormas KA, Maignien L, Olu K, Pachiadaki M, Ritt B, Rodrigues C, Sarrazin J, Tyler P, Van Gaever S, Vanneste H (2009) Biodiversity of cold seep ecosystems along the European margins. Oceanography 22:110–127. https://doi.org/10.5670/oceanog.2009.12
Kaygorodova IA (2011) Deep-water fauna of Oligochaeta (Annelida, Clitellata) near hydrothermal spring of the Frolikha Bay, Northern Baikal (East Siberia, Russia). J Siberian Federal Univ Biol 2(4):117–132. https://doi.org/10.17516/1997-1389-0175
Levin LA, Michener RH (2002) Isotopic evidence for chemosynthesis-based nutrition of macrobenthos: the lightness of being at Pacific methane seeps. Limnol Oceanogr 47:1336–1345. https://doi.org/10.4319/lo.2002.47.5.1336
Goffredi SK, Tilic E, Mullin SW, Dawson KS, Keller A, Lee RW, Wu F, Levin LA, Rouse GW, Cordes EE, Orphan VJ (2020) Methanotrophic bacterial symbionts fuel dense populations of deep-sea feather duster worms (Sabellida, Annelida) and extend the spatial influence of methane seepage. Sci Adv 6(14):eaay8562. https://doi.org/10.1126/sciadv.aay8562
Jones R, Grey J (2011) Biogenic methane in freshwater food webs. Freshwater Biol 56:213–229. https://doi.org/10.1111/j.1365-2427.2010.02494.x
Jones SE, Lennon JT (2009) Evidence for limited microbial transfer of methane in a pelagic food web. Aquat Microb Ecol 58:45–53. https://doi.org/10.3354/ame01349
Leak DJ, Stanley SH, Dalton H (1985) Implication of the nature of methane monooxygenase on carbon assimilation in methanotrophs. In: Poole RK, Dow CS (eds) Microbial gas metabolism, mechanistic, metabolic and biotechnological aspects. Academic Press, London, pp 201–208
Graham DW, Chaudhary JA, Hanson RS, Arnold RG (1993) Factors affecting competition between type I and type II methanotrophs in continuous-flow reactors. Microb Ecol 25:1–17. https://doi.org/10.1007/BF00182126
Raghoebarsing A, Smolders A, Schmid M, Rijpstra W, Wolters-Arts M, Derksen J, Jetten M, Schouten S, Damsté J, Lamers L, Roelofs J, den Camp H, Strous M (2005) Methanotrophic symbionts provide carbon for photosynthesis in peat bogs. Nature 436:1153–1156. https://doi.org/10.1038/nature03802
Mekhanikova IV, Sitnikova TYa (2014) Amphipods (Amphipoda, Gammaridea) at the Gorevoy Utes oil and methane seep, Lake Baikal. Crustaceana 87:1500–1520. https://doi.org/10.1163/15685403-00003367
Lazo FI (1980) Sulfur geochemistry in the bottom sediments of Lake Baikal. Geochemistry 1:109–115
Friedrich CG, Bardischewsky F, Rother D, Quentmeier A, Fischer J (2005) Prokaryotic sulfur oxidation. Curr Opin Microbiol 8:253–259. https://doi.org/10.1016/j.mib.2005.04.005
Zemskaya TI, Namsaraev BB, Dul’tseva NM, Khanaeva TA, Golobokova LP, Dubinina GA, Dulov LE, Wada E (2001) Ecophysiological characteristics of the mat-forming bacterium Thioploca in bottom sediments of the Frolikha Bay, Northern Baikal. Microbiology 70:335–341
Newton RJ, Jones SE, Eiler A, McMahon KD, Bertilsson S (2011) A guide to the natural history of freshwater lake bacteria. Microb Mol Biol Rev 75:14–49. https://doi.org/10.1128/MMBR.00028-10
Moeller FU, Webster NS, Herbold CW, Behnam F, Domman D, Albertsen M, Mooshammer M, Markert S, Turaev D, Becher D, Rattei T, Schweder T, Richter A, Watzka M, Nielsen PH, Wagner M (2019) Characterization of a thaumarchaeal symbiont that drives incomplete nitrification in the tropical sponge Ianthella basta. Environ Microbiol 21:3831–3854. https://doi.org/10.1111/1462-2920.14732
Vuillemin A, Wankel SD, Coskun ÖK, Magritsch T, Vargas S, Estes ER, Spivack AJ, Smith DC, Pockalny R, Murray RW, D’Hondt S, Orsi WD (2019) Archaea dominate oxic subseafloor communities over multimillion-year time scales. Sci Adv 5:4108. https://doi.org/10.1126/sciadv.aaw4108
Acknowledgements
The State Assignment 0279-2021-0006 supported the work. Microscopic studies were carried out in the Electronmicroscopy center of collective instrumental center “Ultramicroanalysis” Limnological Institute Siberian Branch of the Russian Academy of Sciences (http://www.lin.irk.ru/).
Author information
Authors and Affiliations
Contributions
TZ and TS designed the research and revised the manuscript; SC wrote the manuscript; AK, TS, and TN performed samples; IK did microscopy; SC did molecular analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chernitsyna, S.M., Khalzov, I.A., Sitnikova, T.Y. et al. Microbial Communities Associated with Bentic Invertebrates of Lake Baikal. Curr Microbiol 78, 3020–3031 (2021). https://doi.org/10.1007/s00284-021-02563-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-021-02563-0