
Abstract
Several species of mycobacteria cause infections in humans. Species identification of clinical isolates of mycobacteria is very important for the decision of treatment and in choosing the appropriate treatment regimen. We have developed a multiplex PCR method that can identify practically all known species of mycobacteria, by determination of single-nucleotide differences at a total of 13 different polymorphic regions in the genes of rRNA and hsp65, in four PCR mixes. To achieve this goal, single-nucleotide differences in these polymorphic regions were used to divide mycobacterial species into two groups, than four, eight, etc., in an algorithmic manner. It was sufficient to reach single species level by evaluating 13 polymorphic regions. Evaluation of the multiplex PCR patterns by observable real-time electrophoresis (ORTE) simplified species identification. This new method may enable easy, rapid, and cost-effective identification of all species of mycobacteria.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Tuberculosis continues to be one of the major health problems around the world. Globally, tuberculosis accounts for almost 3 million deaths annually and one-fifth of all deaths of adults in developing countries. Additionally, tuberculosis is a re-emergent problem in many industrialized countries due to rapid transportation, expanding trade, and changing social and cultural patterns [1]. On the other hand, the infections due to non-tuberculosis mycobacteria (NTM) showed a major increase parallel to the rise in the number of immunocompromized patients due to HIV infection, cancer therapy, or chronic diseases like diabetes mellitus [2]. There are more than 170 species of mycobacteria identified so far in nature. NTM are widely found in soil, water, and animals, and can be readily transmitted to humans. They can either cause symptomatic infections or sometimes can be found in human body without causing any disease. Therefore, when mycobacteria are identified in clinical samples it is very important to identify the species, firstly in order to determine if it can be associated with the presenting disease and if it is, to choose the right treatment regimen, since the drugs effective for infections created by different species of mycobacteria vary markedly [3].
Since methods based on culture and biochemical methods for mycobacterial species identification require long time and extensive effort, molecular methods replaced these in recent years. Line probe assay (LIPA) which depends on PCR and reverse hybridization and DNA sequencing are used more and more for mycobacterial species identification. These methods are too cumbersome for resource-limited settings and require expensive equipment and personnel, well trained in molecular methods. Simpler molecular methods for determination of mycobacterial species are a growing necessity [4,5,6].
The main aim of this study was to develop a simple multiplex PCR assay that can determine all known species of mycobacteria by identifying single-nucleotide differences in the genes of different species of mycobacteria. rRNA and hsp65 sequences of mycobacteria that are conserved in some regions and show heterogeneity in other parts have been targeted for this purpose [7]. However, the sequences of these genes are exactly the same in M. tuberculosis, M. bovis, M. africanum, M. microti all of which belong to a group called M. tuberculosis complex [8].
Most of the mycobacterial infections in humans are caused by M. tuberculosis. In many countries around the world, isolation rate of M. tuberculosis-to-NTM ratio is usually over 95%. For this reason, when mycobacteria are isolated in culture, it is logical to first differentiate if it is M. tuberculosis or other mycobacterial species belonging to M. tuberculosis complex or NTM with a PCR specific for only M. tuberculosis. This would identify most of the clinical isolates as M. tuberculosis and only a few isolates would need further evaluation [9]. Therefore, we have designed an additional simple three-reaction PCR panel to differentiate if the isolate is M. tuberculosis or not.
Materials and Methods
Bacterial Strains
Mycobacterial strains were obtained from different culture collections. The source and reference number of 130 strains belonging to different species of mycobacteria, used in this study, are as follows:
M. kansasii (NT 701, ATCC 12478), M. duvalii (NT 4601), M. bovis BCG (ATCC 27289), M. interjectum (DSM 44064, ATCC 51457), M. moriokaense (DSM 44221, ATCC 43059), M. avium subsp silvaticum (DSM 44175, ATCC 49884),M.montefiorense (DSM 44602), M. fürth (DSM44567), M. simiae (NT 909, ATCC 25275), M. chubuense (NT 4701, ATCC 27278), M. kansasii (IMET 10659), M.mucogenicum (DSM 44124), M. murale (DSM 44340),M. botniense (DSM 44537, ATCC 700702), M. nebraskense (DSM 44803), M.fouranthenivorans (DSM44556), M. scrofulaceum (NT 1001, ATCC 19981), M. aichiense (NT 4801, ATCC 27280), M. gordonae (SN 601), M. conspicuum (DSM 44136),M. obuense (DSM 44075, ATCC 27023),M. doricum (DSM 44339), M. neworleansense (DSM 44679),M.hackensackense (DSM44833, ATCC BAA-823), M. gordonae (NT 1101, ATCC 14470), M. tokaiense (NT 4901), M. intracellulare (ATCC 13950), M. shimoidei (DSM 44152, ATCC 27962),M.septicum (DSM 44393, ATCC 700731), M. frederiksbergense (DSM 44346),M.palustre (DSM 44572), M. holsaticum (DSM44478), M. szulgai (NT 1201, ATCC35799), M. gadium (NT 5001, ATCC 27726), M. phlei (ATCC 11758), M. malmoense(DSM 44163), M. tusciae (DSM 44338), M. gilvum (DSM 44503, ATCC 43909),M. parascrofulaceum (DSM 44648), M. kumamotonense (DSM45093), M. gastri (NT 1301,ATCC 15754), M. rhodesiae (NT 5201), M. terrae (ATCC 15755),M.thermoresistibile (DSM 44167,ATCC 19527), M. vaccae (ATCC 29678),M.neoaurum (DSM 44074, ATCC 25795), M. parmense (DSM 44,53), M. manitobense (DSM44615), M. nonchromogenicum (NT 1401, ATCC 19530), M. austroafricanum (NT 5301, ATCC 33464), M.tuberculosis (14323/RIVM), M. abscessus (DSM 44196,ATCC 19977), M.elephantis (DSM 44368), M. bönickei (DSM 44677,M.psychrotolerans (DSM 44697), M. massiliense (DSM45103), M. terrae (NT 1501, ATCC 15755), M. fallax (NT 5501,ATCC 35219), M. celatum (ATCC 51130),M.novocastrense (DSM 44203), M. hodleri (DSM 44183), M. brisbanense (DSM 44680),M. pyrenivorans (DSM 44605), M. monacense (DSM44395), M. triviale (NT 1601, ATCC 23292), M. gallinarum (NT 5601, ATCC 19710), M. celatum (DSM44243, ATCC 51131), M. marinum (DSM 44344, ATCC 927), M. heckeshornense (DSM 44428),M. canariasense (DSM 44828), M. saskatchewanense (DSM 44616), M.neglectum (DSM44756), M. xenopi (NT 1901, ATCC 19250), M. porcinum (NT 5801, ATCC 33775), M. heidelbergense (ATCC 51253), M. alvei (DSM 44176, ATCC 51304), M. wolinskyi (DSM 44493, ATCC 700010), M. cosmeticum (DSM 44829), M. vanbaalenii (DSM 07251), M.paraseoulense (DSM45000), M. vaccae (NT 2201, TMC 1526), M. sphagni (DSM44076, ATCC 33027), M. branderi (DSM44624, ATCC 51789), M. bohemicum (DSM 44277), M. hiberniae (DSM 44241, ATCC 49874), M. florentinum (DSM 44852),M.angelicum (DSM45057), M. petroleophilum (DSM44182, ATCC 21497), M. diernhoferi (DSM43524, ATCC 19340), M. asiaticum (NT 6101, ATCC 25276), M. triplex (DSM 44626), M. brumae (DSM 44177, ATCC 51384), M. mageritense (ATCC 700351),M. haemophilum (DSM 44634), M. arupense (DSM44942, ATCC BAA-1242),M. phocaicum (DSM45104), M. smegmatis (NT 2433,ATCC 14468), M. senegalense (NT 6801), M. flavescens (DSM 43219), M. farcinogenes (DSM 43637,ATCC 35753),M.cookii (DSM 43922, ATCC 49103), M. houstonense (DSM 44676),M.barombii(DSM45059),M. ratisbonense (DSM44364), M. parafortuitum (DSM43528, ATCC 19686), M. pulveris (NT 7001, ATCC 35154), M. peregrinum (DSM 43271, ATCC14467), M. hassiacum (DSM 44199), M. goodii (DSM 44492, ATCC 700504), M. immunogenum (DSM 44764),M. chimaera (DSM44623), M. salmoniphilum (DSM43276, ATCC 13758), M. aurum (NT 4101, ATCC 23366), M. fortuitum (ATCC 6841), M. confluentis (DSM 44017, ATCC 49920), M. komossense (DSM 44078, ATCC 33013), M. tuberculosisH37RV (CDC), M. kubicae (DSM 44627), M. conceptionense (DSM45102), M. seoulense (DSM44998), M. chitae (NT 4301) M. chelonae (DSM43804, ATCC 35752), M. intermedium (DSM 44049, ATCC 51848), M. lentiflavum (DSM 44418, ATCC 51985), M. agri (DSM 44515, ATCC 27406), M. lacus (DSM 44577),M. concordense(DSM44678, ATCC BAA-329),M. setense (DSM45070), M. Yunnanensis (DSM44838), M.senuense (DSM44999).
Bacterial Culture and Isolation of DNA
Mycobacterium DNA isolation was performed by using the boiling method as explained before [10, 11]. Mycobacteria stock cultures stored in − 85 °C were thawed at room temperature and then plated on the Löwenstein–Jensen media. Several colonies from fresh cultures were suspended in 750 µl of TE buffer (10 mM Tris pH 8.0, 1 mM EDTA) in 1.5 ml microcentrifuge tubes. The tubes were centrifuged at 12.000×g for 2 min. The supernatant was discarded and the sediment was resuspended in 750 µl of TE buffer. After centrifugation, supernatant was discarded and the pellets were dissolved in 200 µl of TE. Then, tubes were incubated for 20 min in a water bath at 95 °C [10, 11], since the cell wall structure of the Mycobacteria is composed of durable long fat chains. The tubes were spinned again at 12.000×g for 2 min and the supernatant, which contained mycobacterial DNA, was transferred into a clean tube. The DNA samples were kept at − 20 °C until used for PCR assays.
Differentiation of M. Tuberculosis From Other Species of Mycobacteria
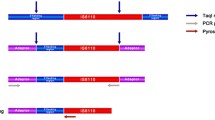
PCR panel, that we named panel A, which differentiated M. tuberculosis from other members of M. tuberculosis complex and NTM, consisted of three reactions. First reaction, A1, shows that this sample belongs any species of mycobacteria. The second reaction, A2, produced a PCR product if the isolate is M. tuberculosis. The third reaction tube A3, contained exactly the same reagents as the first tube but no template DNA, was used as negative control to show that the reaction is completed without any contamination. A1 amplified rRNA gene common to all mycobacteria with primers (RRFC: 5′GGCGTGCTTAACACATGCAAGTCG3′ and RR1F: 5′CCTACGAGCTCTTTACGCCCAGTA3′). A2 amplified a region of gyrB and the primers (MtbGyrBF: 5′GCCGAAGTCGGCAAGTGAACGC3′ and MtbGyrBR1: 5′AGCCTTGTTCACAACGACTTTCGC 3′) only matched M. tuberculosis DNA sequences. PCR amplification results from M. tuberculosis and other species of mycobacteria are shown in Fig. 1.

PCR amplification products obtained by Panel A, which differentiates M. tuberculosis from all other mycobacterial species, including the ones like M. bovis that belong to M. tuberculosis complex. (M: Molecular weight marker − 100 bp ladder; A1: Amplification product by ribosomal RNA primers specific for mycobacteria; A2: Amplification product by gyrB primers specific to M. tuberculosis; A3: Negative control.)
Selection of the Polymorphic Sites for Species Identification
The sequences of rRNA and hsp65 of all mycobacteria available in GenBank were downloaded and aligned by CLC program (http://en.bio-soft.net/format/CLC.html). Five polymorphic regions in rRNA and 8 polymorphic regions in hsp65, which can be used in differentiation of species from each other, were identified. The polymorphic region, which can divide mycobacterial species into almost two equal groups (one type of nucleotide in one group and another type of nucleotide in the other group), was selected as the first target for PCR reaction that can identify this specific nucleotide difference. The second PCR target was the polymorphic region, which can now divide the first two groups one more time into almost two equal groups to divide the whole mycobacterial species into four groups. This algorithm was continued to group the species into eight groups by a third polymorphic region than 16 and so on until all the species can be differentiated individually. If it would be possible to divide species into exactly two equal groups by each analyzed region, analysis of 8 polymorphic regions would theoretically provide 256 combinations and suffice to differentiate all mycobacterial species known so far. However, since each polymorphic region could not divide species into two equal groups, we needed to use 13 polymorphic regions to differentiate all mycobacterial species. The name of the primers used for amplification of these regions, the size of PCR amplification products obtained, and examples of expected product amplification results from different species of mycobacteria, which made our Panel B, are shown in Table 1.
Primer Design for High-Specificity Identification of Polymorphism by PCR
For identification of single-nucleotide differences, primers for PCR were designed to match their 3′ prime ends to the polymorphic nucleotide. We aimed to obtain a PCR product if there was a match at the 3′ end of the primer and no product if there was a mismatch. Since standard PCR primers often produce a PCR product, even though there is a mismatch at 3′ end, we designed special primers to weaken the binding of 3′ end, but without loosing their specificity. After trial of several combinations, we replaced six nucleotides by inosine, preceding the seven nucleotides at the 3′ end of the primers, which differentiated best 3′ end matched and unmatched nucleotides. The primers used in this study are listed in Table 2. For some amplification reactions, we used a standard primer (like RRFC) for polymerization on one direction and an inosine-containing primer for the reverse direction and for others we used inosine-containing primers for both directions.
Multiplex PCR Assay and Electrophoresis
To simplify the application, we have combined three or four reactions among the 13 reactions defined above, into four PCR mixes. Combined reaction in each tube was chosen according to the sizes of the amplification products so that the products can easily be separated from each other by agarose gel electrophoresis. Examples of products amplified from different species of mycobacteria in four multiplex PCR reactions are shown in Fig. 2. Mixes of primers that were used in each multiplex PCR reactions were prepared, put in 0.2 mL PCR tubes, dried in a class II biosafety cabinet, and kept at room temperature until being used for PCR. For PCR assays, 100 µl of mix, including 10 µl of mycobacterial DNA isolated as described above, was prepared and 25 µl was distributed into four multiplex PCR tubes containing the dried primers. The mixes were prepared with a high-fidelity Taq polymerase (Dream Taq—Thermo Fisher Scientific) according to the instruction of the manufacturer using the 10x buffer, which makes the final concentration of Mg++ in the mix 2 mM. Amplification was carried out by 2 min denaturation at 95 °C followed by 45 cycles of 30 s at 95 °C and 10 s at 64 °C. After amplification, PCR products were mixed by SYBR Gold dye and loaded on 2% agarose gel. The gel was placed in ORTE (Observable Real-Time Electrophoresis, TiBO, Istanbul, Turkey) that we have developed previously [12] and electrophoresis was run for 45 min at 100 V by continuous monitoring and taking pictures every 3 min.

Multiplex PCR amplification products from different species of mycobacteria: M: molecular weight marker, 100 bp ladder. Columns 1 to 4: Amplification products of multiplex PCR. The images are obtained by ORTE
Automatic Species Identification of Mycobacterial Species by Pattern Recognition Program of ORTE
We have added a pattern recognition program to the image analysis software of ORTE instrument. ORTE is a real-time electrophoresis system that takes and saves pictures at intervals as desired. At the end of the electrophoresis run, one of the pictures, which showed optimum separation of DNA molecules, was analyzed by pattern recognition software. The program evaluates the sizes of PCR products according to the molecular weight marker, which should be loaded to the gel together with multiplex PCR products. This software is a self-learning program and once the multiplex PCR product pattern from a species of mycobacteria is introduced to the system it can identify the same species of mycobacteria when it evaluates the same pattern.
Results
We have studied 130 species of mycobacteria, listed in Table 1, that were available in our culture collection, using Panel A and Panel B. With Panel A, it was possible to confirm that all strains studied belonged to mycobacteria and to differentiate M. tuberculosis from all other mycobacterial species, including the ones which belonged to M. tuberculosis complex.
Panel B produced distinct multiplex PCR amplification patterns for all 130 mycobacterial species. Once these patterns were recorded in ORTE software to create a library, the instrument correctly identified the species when the strains were retested. The analysis of 13 different single-nucleotide polymorphic sequences in rRNA and hsp65 in an algorithmic way was sufficient to differentiate all species of mycobacteria from each other.
In order to achieve very specific identification of single-nucleotide differences, firstly, standard primers were used. The compatibility of 3′ end nucleotides of these primers to nucleotides on binding sites of template DNA molecules, was investigated by obtaining or not a PCR product which depends on specific priming and initiation of DNA polymerization. Although, several PCR conditions were tried, it was not possible to obtain specificity with standard primers. In many cases, a PCR product was obtained even though the 3′ end of the primer did not match exactly to template DNA. It was possible to obtain specificity by a combination of several changes in the application.
Our first approach was to redesign the primers. It was aimed to release the primers’ 3′ end when there was a mismatch, so that it cannot start polymerization. To weaken the binding of the 3′ end, the original nucleotides of the primer close to the 3′ end were exchanged, by inosines which do not make hydrogen bonds with the template. Different primers were designed by adding 5 to 8 inosines with a distance of five to eight nucleotides away to 3′ end. The best results were obtained with primers having seven nucleotides at 3′ end and 6 inosines preceding these. When inosines were included in primers, it was also observed that at least another 18 nucleotides adjacent to inosines were required at 5′ end of the primer, for proper binding.
Our second approach was to use a high-fidelity enzyme, Dream Taq polymerase, which has proof reading activity that lacks in standard Taq polymerase, which increased specificity of the PCR reactions.
Our third approach was to lower the speed of Taq polymerase by lowering polymerization temperature from 72 to 64 °C, which eliminated several non-specific amplifications.
Discussion
M. tuberculosis is responsible for the majority of mycobacterial infections and it is the leading pathogen, which is responsible for the highest number of patients and deaths around the world as a single infectious agent [13]. The most important measure to control tuberculosis in the community is to diagnose early, the patients who have tuberculosis bacilli in their sputum and treat them efficiently before they transmit the disease to healthy people. On the other hand, parallel to the increase in the number of immunosuppressed patients, the number of infections due to NTM is also increasing, primarily in industrialized countries. Since the treatment regimens of NTM infections vary greatly and different than the infections due to M. tuberculosis, species identification is critical once mycobacteria are identified in clinical samples [14]. Species identification is also important for the decision of treat or not to treat, since several NTM may be found in clinical samples without causing any disease [15].
The number of mycobacterial species detected so far in nature is more than 170 and increasing with identification of new species every year. At least twenty of these can cause infections in humans [3, 15].
Classical way to identify the species of mycobacteria, as suggested by Rounyon [16], depends on growth time on culture, pigment formation of colonies in dark or exposed to light, and different biochemical properties. However, this type of identification requires long time in slow-growing mycobacteria and is very hard to apply [17]. For that reason, phenotypic methods depending on chromatic separation of mycolic acids by HPLC, GLC, or TLC have been developed. Although these methods speed up the species identification, they suffer from being cumbersome and inefficient in differentiating all mycobacterial species from each other [18, 19]. Rapid species identification using Matrix-Assisted Laser Desorption Ionization-Time of Flight (MALDI-TOF) seem to have great promise; however, it requires the presence of this expensive instrument [20]. In the last few decades, several molecular methods for species identification of mycobacteria have been developed. All of these methods depend on sequence differences in certain genes like rRNA and hsp65 [21]. The most specific way is to sequence these genes. However, DNA sequencing is still an expensive and cumbersome method for resource-limited settings. Manual methods like PCR restriction enzyme analysis (PRA) are hard to apply and require experienced personnel. One of the most widely used molecular methods, Line Probe Assay (LIPA), depends on reverse hybridization of PCR-amplified products to specific sequences of different species. Since LIPA makes use of specific DNA probes that match to genes of certain species of mycobacteria, the number of mycobacteria, which can be identified by this test, is limited to the probes included in the test strips [22,23,24].
In this study, we aimed to identify the species of mycobacteria by determination of single-nucleotide polymorphism by PCR. Since standard PCR cannot usually differentiate single-nucleotide differences, several approaches have been previously developed to make PCR identify these differences. Germer et al. used a method which they called Tm shift genotyping, combined allele-specific PCR with the discrimination between amplification products, by their melting temperatures (Tm). Two distinct forward primers, each of which contains a 38-terminal base that corresponds to one of the two SNP allelic variants, were combined with a common reverse primer in a single-tube reaction. A GC-tail was attached to one of the forward allele-specific primers to increase the Tm of the amplification product from the corresponding allele. PCR amplification, Tm analysis, and allele determination of genomic template DNA, were carried out on a fluorescence-detecting thermocycler with a dye that fluoresces when bound to dsDNA. A truncated form of Taq DNA polymerase, Stoffel DNA polymerase, was used in this study. Stoffel fragment has been shown to enhance discrimination of 38 primer–template mismatches where Taq polymerase did not discriminate well, mismatches of T with G, C, or A [25].
In 2007, Chun et al. used a method, which they reported as a novel dual priming oligonucleotide (DPO) which contains two separate priming regions joined by a polydeoxyinosine linker [26]. The linker assumes a bubble-like structure which itself is not involved in priming, but rather delineates the boundary between the two parts of the primer. This structure results in two primer segments with distinct annealing properties: a longer 5′ segment that initiates stable priming, and a short 3′ segment that determines target-specific extension. This DPO-based system is a fundamental tool for blocking extension of non-specifically primed templates, and thereby generates consistently high PCR specificity even under less than optimal PCR conditions. They evaluated the DPO-based system in a multiplex PCR application for the detection of five different human respiratory viruses. In their study, long conventional primers generated many non-specific bands, most likely due to non-specific annealing or primer competition. In contrast, the DPO primer generated target-specific viral fragments, and no false positives [26].
In this study, we have used a similar approach but we optimized our primers for our assay and showed that primers that have six inosines preceding seven specific nucleotides at 3′ end and presence of at least 18 nucleotides at 5′ end preceding inosines distinguished best single-nucleotide differences matching the 3′ end of the primer. Using a high-fidelity Taq polymerase which had proof reading activity and lowering the polymerization temperature from 72 to 64 °C, to decrease the chance for the enzyme to make mistakes, were crucial to obtain the desired specificity.
One of the important achievements of our method was identification of all tested mycobacterial species by analyzing only 13 polymorphic sites in an algorithmic manner. It was possible to determine polymorphisms in these 13 sites by 4 multiplex PCR reactions. We were able to differentiate 130 mycobacterial species from each other by this method.
In our study, we are able to differentiate all mycobacteria species level including Mycobacterium tuberculosis complex strains found in our culture collection with the multiplex PCR panel. However, we are planning to test this multiplex PCR panel method with more clinical samples for making standard in clinical applications.
Once the reliability of this multiplex PCR method that can identify single-nucleotide differences in mycobacterial genes with high fidelity is proved by further trials with clinical strains, it may be a practical tool for mycobacterial species identification for any laboratory that can do PCR and agarose gel electrophoresis. The use of ORTE and its pattern recognition software makes easy the evaluation of multiplex patterns, for mycobacterial species identification.
References
Bloom BR (1994) Tuberculosis Pathogenesis, Protection, and Control, ASM Press, Washington
Sezgin E, Van Natta M, Thorne J, Puhan M, Jabs D (2018) Secular trends in opportunistic infections, cancers and mortality in patients with AIDS during the era of modern combination antiretroviral therapy. HIV Med 19:411–419
Gagneux S (2018) Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol 16:202
Driesen M, Kondo Y, de Jong BC, Torrea G, Asnong S, Desmaretz C, Mostofa KSM, Tahseen S, Whitfield MG, Cirillo DM, Miotto P, Cabibbe AM, ve Rigouts L (2018) Evaluation of a novel line probe assay to detect resistance to pyrazinamide, a key drug used for tuberculosis treatment. Clin Microbiol Infect 24:60–64
REPORT ET (2018) Handbook on tuberculosis laboratory diagnostic methods in the European Union
Betty A, Forbes GSH, Melissa B, Miller SM, Novak M-C, Rowlinson M, Salfinger A, Somoskövi DM, Warshauer ML, Wilson (2018) Practice Guidelines for Clinical Microbiology Laboratories: Mycobacteria, ASM, Washington
PrasadMyneedua AKK (2018) Amplification of Hsp 65 gene and usage of restriction endonuclease for identification of non tuberculous rapid grower mycobacterium, Indian J Tuberc 65(1):57–62
Kim J-U, Ryu D-S, Cha C-H, ve Park S-H (2018) Paradigm for diagnosing mycobacterial disease: direct detection and differentiation of Mycobacterium tuberculosis complex and non-tuberculous mycobacteria in clinical specimens using multiplex real-time PCR. J Clin Pathol 71(9):774–780
Ryan S, Sadda S, Schachat A, Wilkinson C, Schachat A, Hinton D, Wiedemann P, Wiedemann P (2013) Mycobacterial infections, retina. Elsevier, Amsterdam. ISBN: 9781455707379
Kocagöz T, Özkara EY,Ş, Kocagöz S, Hayran M, Sachedeva M, Chambers HF (1993) Detection of Mycobacterium tuberculosis in sputum samples by polymerase chain reaction using a simplified procedure, J Clin Microbiol 31(6):1435–1438
Mozioğlu E, Tamerler AM, Kocagöz C, ZT (2014) A simple guanidinium isothiocyanate method for bacterial genomic DNA isolation, Turk J Biol 38(1):125–129
Kocagöz T (2013) Real-time observable electrophoresis system, which does not require any kind of light filter, Patent, US2013112562 (A1)
Gilpin C, Korobitsyn A, Migliori GB, Raviglione MC, ve Weyer K (2018) The World Health Organization standards for tuberculosis care and management. Eur Respir J 51:1800098
Fariz Nurwidya DH, Burhan E, Yunus F (2018) Molecular Diagnosis of Tuberculosis, Chonnam Medical Journa
Haworth CS, ve Floto RA (2017) Introducing the new BTS guideline: management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 72:969–970
Forbes BA, Hall GS, Miller MB, Novak SM, Rowlinson M-C, Salfinger M, Somoskövi A, Warshauer DM ve Wilson ML (2018) Practice guidelines for clinical microbiology laboratories: mycobacteria. Clin Microbiol Rev 31:e00038–e00017
Balows A, Hausler WJ, Herrmann JR, Kenneth L, Isenberg HD, Shadomy HJ,, (1991). Manual of clinical microbiology, ASM Press, Washington
Butler WR, Cage G, Desmond E, Duffey PS, Floyd MM, Gross WM, Guthertz LS, Jost WM, Ramos LS, Silcox V, Thibert L, Warren N,, (1996). Standardized Method for HPLC Identification of Mycobacteria, U.S Department of Health and Human Services
Frazier KS, Hines ME,, (1993). Differentiation of mycobacteria on the basis of chemotype profiles by using matrix solid-phase dispersion and thin-layer chromatography Clin Microbiol Infect 31(3):610–614
Novakova MNVKS (2017) Identification of Mycobacterium Species by MALDI-TOF Mass Spectrometry, ed. Pulm Care Clin Med 37–42
Acosta FL, Ruiz Serrano MJ, Marín M, Kohl TA, Lozano N, Niemann S, Valerio M, Olmedo M, Pérez-Granda MJ, Pérez MP, Bouza E, Muñoz P, de Viedma DG (2018) Fast update of undetected Mycobacterium chimaera infections to reveal unsuspected cases, J Hosp Infect 100(4):451–455
Olaru ID, Kranzer K, Perera N (2018) Turnaround time of whole genome sequencing for mycobacterial identification and drug susceptibility testing in routine practice, Clin Microbiol Infect 24(6):659–e5
Broda A, Nikolayevskyy V, Casali N, Khan H, Bowker R, Blackwell G, Patel B, Hume J, Hussain W, ve Drobniewski F (2018) Experimental platform utilising melting curve technology for detection of mutations in Mycobacterium tuberculosis isolates. Eur J Clin Microbiol Infect Dis 37:1273–1279
Tortone C, Zumárraga M, Gioffr A, ve Oriani D (2018) Utilization of molecular and conventional methods for the identification of nontuberculous mycobacteria isolated from different water sources. Int J Mycobacteriol 7:53–60
Germer S, Higuchi R, Holland MJ (2000) High-Throughput SNP Allele-Frequency Determination in Pooled DNA Samples by Kinetic PCR, Genome Res 10(2):258–266
Dae-Hoon L, Kyoung I, In-Taek HL, Jong-Kee K, Jong-Yoon C, Kyoung-Joong K, Yun-Jee K, (2007) Dual priming oligonucleotide system for the multiplex detection of respiratory viruses and SNP genotyping of CYP2C19 gene, Nucleic Acids Res 35(6):e40
Acknowledgements
This work was supported by TUBİTAK 107S014 (SBAG-3541) Identification of Mycobacterial Species by Sequence-Specific Polymerase Chain Reaction; for financial aid, I wish to thank TÜBİTAK.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Unubol, N., Kizilkaya, I.T., Okullu, S.O. et al. Simple Identification of Mycobacterial Species by Sequence-Specific Multiple Polymerase Chain Reactions. Curr Microbiol 76, 791–798 (2019). https://doi.org/10.1007/s00284-019-01661-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-019-01661-4