
Abstract
Foodborne disease caused by Vibrio parahaemolyticus is a serious public health problem in many countries. Molecular typing has a great scientific significance and application value for epidemiological research of V. parahaemolyticus. In this study, a real-time PCR with melting curve analysis was established for molecular typing of V. parahaemolyticus. Eighteen large variably presented gene clusters (LVPCs) of V. parahaemolyticus which have different distributions in the genome of different strains were selected as targets. Primer pairs of 18 LVPCs were distributed into three tubes. To validate this newly developed assay, we tested 53 Vibrio parahaemolyticus strains, which were classified in 13 different types. Furthermore, cluster analysis using NTSYS PC 2.02 software could divide 53 V. parahaemolyticus strains into six clusters at a relative similarity coefficient of 0.85. This method is fast, simple, and conveniently for molecular typing of V. parahaemolyticus.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vibrio parahaemolyticus is a Gram-negative halophilic bacterium that thrives in seawater and seafood, and eating food contaminated by this bacterium can cause food poisoning. In coastal countries such as China [1], the United States [2], and Japan [3], this organism has become a crucial foodborne pathogen. In China’s coastal provinces, most of bacterial food poisoning cases are caused by V. parahaemolyticus. With the increase in reported cases of food poisoning caused by V. parahaemolyticus, its epidemiological investigation has attracted more and more attention. Bacterial typing is an important method of epidemiological research and it is of great scientific significance and application value to judge the genetic relationship between strains, trace the source of pathogens, cut off the route of transmission, and draw up effective countermeasures.
Currently, the typing methods of V. parahaemolyticus can be classified into two types: phenotypic typing and molecular typing. The method of phenotypic typing has been unable to meet the needs of V. parahaemolyticus epidemiological research because it contains tedious and time-consuming operations. Furthermore, these operations require experience for interpretation and are limited by subjectivity and low specificity. In recent years, with the development of molecular biology technology, molecular typing assays have played an increasingly important role in epidemiological research, such as pulsed-field gel electrophoresis (PFGE), multilocus sequencing typing (MLST) and multilocus variable number tandem repeat analysis (MLVA), random amplified polymorphic DNA (RAPD), ribotyping, arbitrarily primed PCR (AP-PCR), amplified fragment length polymorphism (AFLP), and enterobacterial repetitive intergenic consensus-PCR (ERIC-PCR) [4,5,6]. Compared with the phenotypic typing method, these methods have advantages of rapid and high specificity, but they all are still laborious because of the need for electrophoretic analysis.
Real-time PCR can detect samples by monitoring the fluorescence changes and has advantages of simple operation and high degree of automation due to no tedious electrophoretic analysis. It has been widely used in the detection of V. parahaemolyticus and other pathogens [7,8,9], but there is too little research on molecular typing using real-time PCR. Genomic polymorphisms analysis and epidemiological research of V. parahaemolyticus by microarray-based comparative genome hybridization (M-CGH) have disclosed 18 large variably presented gene clusters (LVPCs), which is variably distributed within the genomes of different V. parahaemolyticus strains [10, 11]. Xiao et al., applied these 18 LVPCs to group 251 global strains of V. parahaemolyticus using conventional PCR and gel electrophoresis and the results demonstrated checking LVPCs presence or absence gave a resolution for discriminating V. parahaemolyticus strains [12]. The aim of this study was to establish a method by using real-time PCR to check LVPCs presence or absence for molecular typing of V. parahaemolyticus.
Materials and Methods
Bacterial Strains
A total of 53 V. parahaemolyticus strains were used in this study, all strains were isolated by our laboratory from food or patients with diarrhea in Jiaxing, China (Table 1) and confirmed by biochemical characterization. All strains were cultured and maintained on thiosulfate citrate bile salts sucrose (TCBS) plates. Genomic DNA of V. parahaemolyticus strains was extracted with KAPA Express Extract Kits (KAPA Biosystems, Woburn, USA) according to the manufacturer’s instruction.
Oligonucleotide Primers
Sequences of 18 LVPCs were downloaded from Genbank (http://www.ncbi.nlm.nih.gov/Genbank/GenbankSearch.html) and primers of target genes were designed using Primer Premier 5.0 (Premier Biosoft International, Palo Alto, CA, USA). The sequences of primer pairs of target LVPCs used in this study are shown in Table 2.
Optimization of Multiplex Real-Time PCR
Primer pairs of 18 LVPCs were distributed into three tubes and the distribution of primer pairs is also listed in Table 2. The multiplex real-time PCR was optimized by varying a single parameter while other parameters were maintained constant. Briefly, the parameters evaluated include target primer concentration from 0.1 to 0.8 µM and annealing/extension temperature from 55 to 65 °C.
Multiplex real-time PCR was performed in a 20 µl reaction mixture for each tube containing 1 µl DNA template, 3 µl EvaGreen dye, 10 µl KAPA 2G Fast Multiplex PCR Mix, moderate target primers, and corresponding distilled water. The PCR amplification was performed in a Bio-Rad CFX 96™ real-time PCR system and the program was as follows: 95 °C for 3 min, followed by 30 cycles of denaturation at 95 °C for 15 s, annealing temperature for 30 s, and extension at 72 °C for 5 s. Fluorescence signals were measured after extension step of each cycle.
Melting Temperature Curve Analysis
Fluorescence melting temperature curve analysis was performed after PCR amplification. The PCR products were kept to 77 °C and were then heated to 93 °C at a rate of 0.1 °C/s. Fluorescence signals were continuously monitored during the whole process of melting temperature curve analysis.
Data Analysis
Scores of ‘0’ and ‘1’ were attributed to the absence or the presence of a LVPC, respectively, for data obtained from multiplex PCR amplification and melting temperature curve analysis. A cluster analysis was performed by NTSYS PC 2.02 software (Exeter Software, East Setauket, NY) and the unweighted-pair group method using arithmetic averages (UPGMA).
Results
Optimization of Multiplex Real-Time PCR
In order to develop a multiplex real-time RT-PCR that can identify the presence of V. parahaemolyticus LVPCs, a systematic study was performed to optimize the conditions. The optimal primer combination was 0.5 µM of VP0383, 0.04 µM of VP1091, 0.12 µM of VP1778, 0.25 µM of VP2902, 0.26 µM of VPA0074, and 0.16 µM of VPA0716 for tube 1; 0.12 µM of VP0635, 0.6 µM of VP1393, 0.15 µM of VP1563, 0.18 µM of VPA0895, 0.12 µM of VPA 1336, and 0.18 µM of VPA1708 for tube 2; 0.02 µM of VP 1351, 0.4 µM of VP2132, 0.1 µM of VPA0440, 0.1 µM of VPA1199, 0.26 µM of VPA1256, and 0.4 µM of RPI08 for tube 3. The optimal annealing/extension temperature was 62 °C.
PCR Amplification and Melting Temperature Curve Analysis
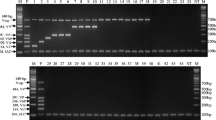
The PCR analyses were performed in five different runs. The VP0383 primer pairs produced PCR products with melting peak at 81.8 ± 0.5 °C, the VP1091 primer pairs produced PCR products with melting peak at 89.4 ± 0.1 °C, the VP1778 primer pairs produced PCR products with melting peak at 87.1 ± 0.3 °C, the VP2902 primer pairs produced PCR products with melting peak at 84.1 ± 0.3 °C, the VPA0074 primer pairs produced PCR products with melting peak at 88.5 ± 0.3 °C, the VPA0716 primer pairs produced PCR products with melting peak at 85.6 ± 0.4 °C, the VP0635 primer pairs produced PCR products with melting peak at 87.4 ± 0.4 °C, the VP1393 primer pairs produced PCR products with melting peak at 85.5 ± 0.5 °C, the VP1563 primer pairs produced PCR products with melting peak at 84.1 ± 0.6 °C, the VPA0895 primer pairs produced PCR products with melting peak at 90.5 ± 0.1 °C, the VPA1336 primer pairs produced PCR products with melting peak at 81.3 ± 0.4 °C, the VPA1708 primer pairs produced PCR products with melting peak at 89.5 ± 0.2 °C, the VP1351 primer pairs produced PCR products with melting peak at 89.3 ± 0.2 °C, the VP2132 primer pairs produced PCR products with melting peak at 81.6 ± 0.6 °C, the VPA0440 primer pairs produced PCR products with melting peak at 86.9 ± 0.4 °C, the VPA1199 primer pairs produced PCR products with melting peak at 88.3 ± 0.4 °C, the VPA1256 primer pairs produced PCR products with melting peak at 83.3 ± 0.5 °C, and the RPI08 primer pairs produced PCR products with melting peak at 85.3 ± 0.6 °C (Fig. 1). All PCR products were further confirmed by gel electrophoresis (Online Resource 1).


Melting peaks of 18 LVPCs obtained after PCR amplification. a Melting peaks produced by primer pairs in tube 1, b melting peaks produced by primer pairs in tube 2, and c melting peaks produced by primer pairs in tube 3
Analysis of V. parahaemolyticus Strains
Fifty-three V. parahaemolyticus strains were tested by the newly developed assays. Genotyping of tested V. parahaemolyticus strains with PCR amplification and melting temperature curve analysis revealed 13 different types. Figure 2 showed the analysis results of VP23. Furthermore, cluster analysis using NTSYS PC 2.02 software could classify 53 V. parahaemolyticus strains into six clusters at a relative similarity coefficient of 0.85 (Fig. 3).

Molecular typing of Vibrio parahaemolyticus strain VP23. a Melting peaks produced in tube 1, b melting peaks produced in tube 2, and c melting peaks produced in tube 3

Dendrogram of the 53 Vibrio parahaemolyticus strain
Discussion
In many countries, intestinal infectious diseases caused by V. parahaemolyticus is a serious public health problem and need to be primarily monitored; virulence gene detection and molecular typing analysis in V. parahaemolyticus isolates are important parts of the monitoring work [13,14,15]. Because real-time PCR was applied in virulence genes detection [16,17,18], the degree of automation for virulence gene detection has been markedly improved. However, the molecular typing analysis for V. parahaemolyticus isolates still uses some traditional methods that are commonly dependent on electrophoresis analysis, thus it is low degree of automation, and time-consuming and laborious. Recently, some researchers have already devoted to using real-time PCR to conduct molecular typing analysis of pathogens. For example, Pang et al. performed the molecular typing analysis of Mycobacterium tuberculosis using real-time PCR combined with high-resolution melt (HRM) technology [19]. But the method used in their study requires HRM technology that has many limitations. For example, HRM is very sensitive, however, results of HRM can be influenced by initial nucleic acid template or amplimer concentrations [20]. Moreover, not all real-time PCR instruments are suitable for HRM analysis. Pang et al. [19] recommended the use of Roche 480 to ensure the credibility of experimental results. In this study, we developed a method for molecular typing of V. parahaemolyticus based on conventional resolution melt technology applied in real- time PCR. This method has the advantage that does not require HRM technology, as applied in the study of Pang et al. [16].
In the long evolutionary process, microorganisms obtain new genes through the transfer of genes, and stably preserve new genes that are beneficial to adaptive microevolution in the population under natural selection pressures, for example, V. parahaemolyticus acquires pathogenicity islands (VPAIs) by horizontal gene transfer [11] thereby causing diseases to humans. Meanwhile, V. parahaemolyticus deletes some DNA regions that are not conducive to microbial survival and reproduction and enables it to be stably inherited in the population. Thus, the acquisition and deletion of genes as a main molecular evolution strategy of bacterial genomes greatly increases the genetic polymorphism of V. parahaemolyticus [21]. Previous studies using M-CGH have screened 18 large LVPCs of V. parahaemolyticus [10, 11]. LVPC refers to large fragments of gene clusters containing at least 10 consecutive genes, these large fragments of gene clusters have different distributions in the genomes of different strains, for example, they are absent in some strains but present in other strains. The study of Xiao et al. has shown that genomic analysis of V. parahaemolyticus based on LVPCs is a very effective method for the genotyping of V. parahaemolyticus [12]. However, the analysis of V. parahaemolyticus LVPCs in their study still depended on the conventional PCR and gel electrophoresis, that was time-consuming and labor-intensive. In this study, we analyzed the LVPCs of V. parahaemolyticus by using real-time PCR combined with melting curve analysis, determining whether the corresponding LVPCs existed according to the presence or absence of melting curve peak. As a result, it eliminates the cumberse electrophoresis process, compared with the method used by Xiao et al., this method is simple, and time- and effort-saving. In addition, it is different from the method used by Pang et al., which used the HRM technique to analyze the molecular typing analysis based on the changes of HRM profiles. We use conventional resolution melt technology and conduct molecular typing analysis according to the presence or absence of melting curve peak, thus, the results are not influenced by initial nucleic acid template or amplimer concentrations and it could be applied in most commercially available real-time PCR instruments.
In conclusion, the method developed by us is fast, simple, and practical, in view of its advantages, it has a potential application as a useful tool for tracing infectiousness and investigating outbreaks caused by V. parahaemolyticus. The advantages of this new method are obvious, but we must admit that there are some aspects that need to be further improved, for example, in order to ensure that the LVPC amplifiers are clearly distinguishable, we have use 6 LVPCs as a group, and 18 LVPCs analysis is performed in 3 different tubes. In the future, we consider establishing a multi-color melting curve analysis method in combination with multi-color labeled probes and melting curve analysis to complete 18 LVPCs detection within a reaction tube.
References
Wang S, Duan H, Zhang W, Li JW (2007) Analysis of bacterial foodborne disease outbreaks in China between 1994 and 2005. FEMS Immunol Med Microbiol 51:8–13. https://doi.org/10.1111/j.1574-695X.2007.00305.x
Iwamoto M, Ayers T, Mahon BE, Swerdlow DL (2010) Epidemiology of seafood-associated infections in the United States. Clin Microbiol Rev 23:399–411. https://doi.org/10.1128/CMR.00059-09
Mahmud ZH, Neogi SB, Kassu A, Wada T, Islam MS, Nair GB, Ota F (2007) Seaweeds as a reservoir for diverse Vibrio parahaemolyticus populations in Japan. Int J Food Microbiol 118:92–96. https://doi.org/10.1016/j.ijfoodmicro.2007.05.009
Lüdeke CH, Gonzalez-Escalona N, Fischer M, Jones JL (2015) Examination of clinical and environmental Vibrio parahaemolyticus isolates by multi-locus sequence typing (MLST) and multiple-locus variable-number tandem-repeat analysis (MLVA). Front Microbiol 6:564. https://doi.org/10.3389/fmicb.2015.00564
Lüdeke CH, Fischer M, LaFon P, Cooper K, Jones JL (2014) Suitability of the molecular subtyping methods intergenic spacer region, direct genome restriction analysis, and pulsed-field gel electrophoresis for clinical and environmental Vibrio parahaemolyticus isolates. Foodborne Pathog Dis 11:520–528. https://doi.org/10.1089/fpd.2013.1728
Sahilah AM, Laila RA, Sallehuddin HM, Osman H, Aminah A, Ahmad Azuhairi A (2014) Antibiotic resistance and molecular typing among cockle (Anadara granosa) strains of Vibrio parahaemolyticus by polymerase chain reaction (PCR)-based analysis. World J Microbiol Biotechnol 30:649–659. https://doi.org/10.1007/s11274-013-1494-y
Heo EJ, Song BR, Park HJ, Kim YJ, Moon JS, Wee SH, Kim JS, Yoon Y (2014) Rapid detection of Listeria monocytogenes by real-time PCR in processed meat and dairy products. J Food Prot 77:453–458. https://doi.org/10.4315/0362-028X.JFP-13-318
Botaro BG, Cortinhas CS, Março LV, Moreno JF, Silva LF, Benites NR, Santos MV (2013) Detection and enumeration of Staphylococcus aureus from bovine milk samples by real-time polymerase chain reaction. J Dairy Sci 96:6955–6964
Liu B, He X, Chen W, Yu S, Shi C, Zhou X, Chen J, Wang D, Shi X (2012) Development of a real-time PCR assay for rapid detection of Vibrio parahaemolyticus from seafood. Protein Cell 3:204–212. https://doi.org/10.1007/s13238-012-2017-6
Okada N, Iida T, Park KS, Goto N, Yasunaga T, Hiyoshi H, Matsuda S, Kodama T, Honda T (2009) Identification and characterization of a novel type III secretion system in trh-positive Vibrio parahaemolyticus strain TH3996 reveal genetic lineage and diversity of pathogenic machinery beyond the species level. Infect Immun 77:904–913. https://doi.org/10.1128/IAI.01184-08
Izutsu K, Kurokawa K, Tashiro K, Kuhara S, Hayashi T, Honda T, Iida T (2008) Comparative genomic analysis using microarray demonstrates a strong correlation between the presence of the 80-kilobase pathogenicity island and pathogenicity in Kanagawa phenomenon-positive Vibrio parahaemolyticus strains. Infect Immun 76:1016–1023. https://doi.org/10.1128/IAI.01535-07
Xiao X, Yan Y, Zhang Y, Wang L, Liu X, Yang L, Tan Y, Guo Z, Yang R, Zhou D (2011) A novel genotyping scheme for Vibrio parahaemolyticus with combined use of large variably-presented gene clusters (LVPCs) and variable-number tandem repeats (VNTRs). Int J Food Microbiol 149:143–151. https://doi.org/10.1016/j.ijfoodmicro.2011.06.014
Mala W, Alam M, Angkititrakul S, Wongwajana S, Lulitanond V, Huttayananont S, Kaewkes W, Faksri K, Chomvarin C (2016) Serogroup, virulence, and molecular traits of Vibrio parahaemolyticus isolated from clinical and cockle sources in northeastern Thailand. Infect Genet Evol 39:212–218. https://doi.org/10.1016/j.meegid.2016.01.006
Tsai SE, Jong KJ, Tey YH, Yu WT, Chiou CS, Lee YS, Wong HC (2013) Molecular characterization of clinical and environmental Vibrio parahaemolyticus isolates in Taiwan. Int J Food Microbiol 165:18–26. https://doi.org/10.1016/j.ijfoodmicro.2013.04.017
Chowdhury G, Ghosh S, Pazhani GP, Paul BK, Maji D, Mukhopadhyay AK, Ramamurthy T (2013) Isolation and characterization of pandemic and nonpandemic strains of Vibrio parahaemolyticus from an outbreak of diarrhea in North 24 Parganas, West Bengal, India. Foodborne Pathog Dis 10:338–342. https://doi.org/10.1089/fpd.2012.1340
He P, Chen Z, Luo J, Wang H, Yan Y, Chen L, Gao W (2014) Multiplex real-time PCR assay for detection of pathogenic Vibrio parahaemolyticus strains. Mol Cell Probes 28:246–250. https://doi.org/10.1016/j.mcp.2014.06.001
Robert-Pillot A, Copin S, Gay M, Malle P, Quilici ML (2010) Total and pathogenic Vibrio parahaemolyticus in shrimp: fast and reliable quantification by real-time PCR. Int J Food Microbiol 143:190–197. https://doi.org/10.1016/j.ijfoodmicro.2010.08.016
Nordstrom JL, Vickery MC, Blackstone GM, Murray SL, De Paola A (2007) Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenic Vibrio parahaemolyticus bacteria in oysters. Appl Environ Microbiol 73:5840–5847. https://doi.org/10.1128/AEM.00460-07
Pang Y, Zhou Y, Wang S, Lu J, Lu B, He G, Wang L, Zhao Y (2011) A novel method based on high resolution melting (HRM) analysis for MIRU-VNTR genotyping of Mycobacterium tuberculosis. J Microbiol Methods 86:291–297. https://doi.org/10.1016/j.mimet.2011.05.016
Tong SY, Giffard PM (2012) Microbiological applications of high-resolution melting analysis. J Clin Microbiol 50:3418–3421. https://doi.org/10.1128/JCM.01709-12
Han H, Wong HC, Kan B, Guo Z, Zeng X, Yin S, Liu X, Yang R, Zhou D (2008) Genome plasticity of Vibrio parahaemolyticus: microevolution of the ‘pandemic group’. BMC Genom 9:570. https://doi.org/10.1186/1471-2164-9-570
Acknowledgements
This study was supported by Science and Technology Program of Jiaxing City (No. 2013AY21051-1 and 2017AY33071).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
He, P., Wang, H., Luo, J. et al. A Real-Time PCR with Melting Curve Analysis for Molecular Typing of Vibrio parahaemolyticus. Curr Microbiol 75, 1206–1213 (2018). https://doi.org/10.1007/s00284-018-1511-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-018-1511-3