
Abstract
Clustered regularly interspaced short palindromic repeats (CRISPR)—CRISPR-associated protein (Cas) is a microbial adaptive immune system. CRISPR-Cas systems are classified into two main classes and six types. Cpf1 is a putative type V (class II) CRISPR effector, which has revolutionized the genome editing approaches through multiple distinct features such as using T-rich protospacer-adjacent motif, applying a short guide RNA lacking trans-activating crRNA, introducing a staggered double-strand break, and possessing RNA processing activity in addition to DNA nuclease activity. In the present review, we attempt to highlight most recent advances in CRISPR-Cpf1 (CRISPR-Cas12a) system in particular, considering ground expeditions of the nature and the biology of this system, introducing novel Cpf1 variants that have broadened the versatility and feasibility of CRISPR-Cpf1 system, and lastly the great impact of the CRISPR-Cpf1 system on the manipulation of the genome of prokaryotic, mammalian, and plant models is summarized. With regard to recent developments in utilizing the CRISPR-Cpf1 system in genome editing of various organisms, it can be concluded with confidence that this system is a reliable molecular toolbox of genome editing approaches.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The clustered regularly interspaced short palindromic repeats (CRISPR)—CRISPR-associated protein (Cas) system is a natural adaptive immunity in most bacteria and archaea. CRISPR-Cas system helps its host to fight against phages and foreign mobile genetic elements, particularly plasmids [3]. In the recent literature, CRISPR-Cas systems are classified into two major classes (classes I and II) and six types (types I–VI), which is sorted into 19 subtypes [45]. CRISPR-Cas9, classified in class II, is the simplest form of type II CRISPR-Cas system that includes three components: mature CRISPR RNA (crRNA), tracrRNA, and Cas9 protein [26]. When host cells are infected by foreign DNAs, the reaction of the CRISPR-Cas9 system is thought to consist of three steps: (i) a part of the invading DNA is integrated into the CRISPR array as a spacer (adaptation), (ii) the CRISPR array is transcribed to make three essential components (expression), and (iii) the crRNA–tracrRNA complex attaches to its commentary sequence on the foreign genome through the crRNA and then recruits Cas9 endonuclease to introduce the double-strand break (DSB) at the target site (interference) [9].
Streptococcus pyogenes CRISPR-Cas9 (SpCas9) instantaneously adapted as a novel genome editing tool in mammalian cells, where two main repair pathways, non-homologous end joining (NHEJ) and homology-directed repair (HDR), are recruited to rectify the introduced DSBs. The simplicity of CRISPR-Cas9 was improved by fusing together the crRNA and the tracrRNA to form a single guide RNA (sgRNA), to be expressed from a single RNA polymerase III promoter [25]. The feasibility and versatility of CRISPR-Cas9 are well presented in multiple studies, including disease modeling [10, 28], Cancer therapy [5], correction of deleterious mutations [41], drug discovery [14], and antiviral therapy [29]. Although CRISPR-Cas9 has shown its great impact on different approaches, off-target mutations are undesirable effects that are compiled into the host genome as well by this system. To minimize the off-target effects, various strategies have been employed including optimizing the sgRNA blueprint and controlling the potency of Cas9 nuclease [3, 7, 39, 53]. The other limitation of CRISPR-Cas9 system is PAM-dependency, which mostly relies on the canonical form 5′-NGG-3′ or its alternative form 5′-NAG-3′ at a low frequency [78]. Three strategies are considered to overcome the PAM-dependency limitation: first, using altered PAM specificity variants of SpCas9 that can recognize alternative PAM sequences through introducing specific mutation in the PAM identifying domain [38]; second, using Cas9 orthologs, which recognize the PAM with different canonical forms such as NNAGAAW (Streptococcus thermophilus), NNNNGATT (Neisseria meningitidis (N. meningitidis)), and NNGRRT (Staphylococcus aureus) [12, 22, 27, 52, 76]; and third, utilizing the other types of CRISPR effectors such as, Cpf1, which identifies a T-rich PAM at the 5′-site of the protospacer [72].
Cpf1: A Novel Implement Based on CRISPR-Cas System
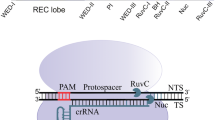
CRISPR-Cpf1 is categorized as type V of class II CRISPR system and was first characterized in Francisella novicida U112 (FnCpf1). FnCpf1 is a large protein, ~1300 amino acids, that is expressed distinctly from the Cas9 locus. The Cpf1 CRISPR array comprises nine spacer sequences which are disassociated by repeated sequences [each 36-nucleotide (nt)]. By comparing 16 Cpf1 orthologs to examine their genome editing activity in human cells, the orthologs from FnCpf1, Moraxella bovoculi (Mb3Cpf1), Lachnospiraceae bacterium (LbCpf1), and Acidaminococcus sp. BV3L6 (AsCpf1) showed promising results that expand the genome editing toolbox. The canonical PAM form for all the orthologs was assumed 5′-TTTV-3′ (V can be C, G, or A), albeit Mb3Cpf1 also represented a recognition activity at shortened PAM (TTN) with low efficiency [71]. CRISPR-Cpf1 possesses four main features: (i) Cpf1 has RNase activity to process its own CRISPR array [15]; (ii) the Cpf1-associated mature crRNA is free of tracrRNA and similar to CRISPR-Cas system (type I or III) includes repeat sequence at 5′- and spacer sequence at 3′-site [6, 20]; (iii) the Cpf1-crRNA complex needs a 5′ T-rich PAM to expeditiously cut the target DNA; and (iv) a staggered DSB (5- or 8-nucleotides 5′-overhang respect to crRNA length) is introduced by Cpf1 orthologs at cleaved sites [43, 72] (Fig. 1).

Schematic illustration of CRISPR-Cpf1 mechanism. A representation of CRISPR-fnCpf1 locus (a). The mechanism of introducing staggered DSB at target site by FnCpf1 (b), red triangles indicate cleavage sites. (Color figure online)
It has been shown that Cpf1 is a monomer in solution and this claim was proven by observing no need to oligomerization for Cpf1 orthologs in crystal structures [1, 8]. Moreover, it is more comprehensible that active Cpf1 is a monomer because if Cpf1 acts in dimer formation, it would demand a tandem DNA target site or otherwise two different crRNAs to target both strands. The RNase activity of Cpf1 on its cognate pre-crRNA was observed to be dependent on repeats and hairpin structure [15]. Cpf1 cuts the 4-nt upstream of the stem-loop and is remindful of the enzymes, Cas6 and Cas5d, which identify the hairpin formation of their repeats [4, 46]. Cpf1 endoribonuclease is metal-dependent and cleaves its RNA target in a structure- and sequence-specific manner. The RNase activity of Cpf1 is increased by addition of Mg2+ because it is unified in the structure of the crRNA which is in contrast with ion-independency of Cas6 and Cas5d [8, 20, 46]. In addition, it has been revealed that introducing mutations such as H843A, K852A, K869A, and F873A can almost obstruct the RNA processing activity but it has no invalidating influence on the RNA-binding activity of FnCpf1 [15, 64]. Furthermore, mutation in conserved residues, (H800A, K809A, K860A, F864A, and R790A) in AsCpf1 also completely disrupts CRISPR array processing but not DNA cleavage activity. To confirm the efficiency of the five variants of AsCpf1, dosage tests were performed and revealed that only the variant harboring, H800A, maintains inactivity in RNA processing regardless of dosage and incubation time, while K809A, K860A, F864A, and R790A variants showed a restoration in their pre-crRNA processing activity at higher dosage or prolonged incubation times. In addition, H800A AsCpf1 was able to disrupt the RNA processing in the HEK293T cell line which also demonstrates that, in mammalian cells, RNA and DNA cleavage activity can be split [73].
Compared to SpCas9, Cpf1 is more sensitive to mismatches within the target site. It has been elucidated that 8-nt at the PAM-proximal site and PAM-distal nucleotides (positions 1–4) at the cleavage site are extremely sensitive to mismatch [15]. Similar to SpCas9, Cpf1 first recognizes the PAM and then explores the 3′-juxtaposed crRNA matching to the target DNA but in a DNA recognition structure distinct from the form reported for SpCas9 [18, 54, 56, 57]. Mismatches around the target site seem to disturb the appropriate binding of Cpf1 and as a result weaken the DNA cleavage activity. The DNA cleavage activity of Cpf1 on both strands of the target site is mediated by a single RuvC-like domain, in contrast to SpCas9 that has two catalytic domains (HNH and RuvC), which cut the complementary and non-complementary DNA strands to guide RNA, respectively [50, 51, 67, 72]. Furthermore, the existing single endonuclease domain in Cpf1 protein was further approved by indicating that there is no difference in cleavage activity of Cpf1 on both complementary and non-complementary DNA strands in the presence of Mg2+ or Ca2+ [15]. These observations were in contrast to the existence of difference in the cleavage activity of SpCas9 on both target strands in the presence of Mg2+ or Ca2+, because SpCas9 has two different catalytic domains that show distinct DNA cleavage efficiency with different ions, for example, the HNH catalytic domain showed Ca2+-dependency in N. meningitidis [25, 75]. Introducing various mutations at different residues in Cpf1 presented differences in DNA cleavage activity at target regions. Mutagenesis in the conserved residues (D917A, D1225A, and E1006A) in the RuvC-like domain of FnCpf1 disrupted the DNA cleavage activity while these mutations had no influence on binding affinity to target DNA or Cpf1 RNA processing strength [15, 72]. Moreover, some mutations have shown differences in the DNA cleavage activity in the presence of different ions. Cpf1 variants, harboring E920A, Y1024A, and D1227A mutations, accompanied by Ca2+ did not cut the target DNA, but they showed wild-type activity in the presence of Mg2+. Additionally, different residues are involved in cutting each target strand, in particular; mutation in the E1028 residue decreased the cleavage of non-complementary strand in the presence of Mg2+, while mutating residues H922 and Y925 in the presence of Ca2+ had shown disruption at the cleavage of complementary strand [15]. In this line, utilizing specific ions in combination with Cpf1 endonuclease variants can help us to arbitrarily cut the desired target strand.
To expand the use of CRISPR-Cpf1 system as a versatile genome editing tool, it is necessary to address its limitation which is the need for the canonical T-rich PAM site. To scale up the targeting range of Cpf1 orthologs, AsCpf1 and LbCpf1, they were subjected to a structure mutagenesis screen. The wild-type AsCpf1 recognizes the TTTV PAM site in the target region, but by introducing the mutations S542R/K548V/N552R (RVR variant) and S542R/K607R (RR variant), which identify TATV and TYCV (Y can be C or T, and V is arbitrarily chosen to be C) PAMs, respectively, the novel engineered variants can keep their robust cleavage activity in vitro and in vivo (RR ~70% and RVR ~78%). The DNA-targeting accuracy of the RR and RVR was assessed using BLISS which indicated the high specificity of these variants at different target regions. Moreover, to improve the specificity of Cpf1 PAM recognition variants, K949A mutation (positioned in the cleft of the Cpf1 protein to interact with the non-complementary strand) was combined with RR and RVR variants, which led to modification reduction at all off-target regions and resulted in higher specificity for both variants. The aforementioned alterations extend the range of PAM recognition of Cpf1 protein and have been predicted to be a commonly befitting approach for Cpf1 orthologs [17, 47]. Likewise, recently, Yamano et al. in a crystal structure study reported that PAM-binding channel of Cpf1, especially LbCpf1, shows conformational flexibility in recognizing canonical (TTTV) and non-canonical (CTTV, TCTV, TTCV) PAMs [66]. Taken together, the engineered AsCpf1 for recognizing altered PAMs and conformational flexibility of the LbCpf1 PAM-binding channel improves our knowledge about the biology of the Cpf1 orthologs and also facilitates the generation of engineered Cpf1 with modified PAM specificities.
Off-target effects and genome editing efficiency are main considerations for utilizing CRISPR systems. Chemical modifications on either crRNA or Cpf1 mRNA have been shown a great impact on improvement of genome editing activity. It has been demonstrated that chemically modified crRNA, having five 2′-fluoro ribose at the 3′ terminus (cr3′5F), enhanced target-cutting efficiency by 127% with respect to wild-type crRNA. Moreover, chemically modified AsCpf1 (full ψ-modification) also maximized gene-cutting efficiency by 177% with respect to plasmid-encoding AsCpf1. Surprisingly, when the ψ-modified AsCpf1 or LbCpf1 were associated with cr3′5F, gene-cutting efficiency was enhanced by 300% in human mammalian cell. According to these findings, it can be concluded that this engineering strategy has a wide pertinency for Cpf1 ortholog-mediated genome editing [44].
Application of CRISPR-Cpf1 System from Bacteria to Mammalian
Corynebacterium glutamicum (C. glutamicum) is a valuable expression system for the production of amino acids and manufacturing biofuels and polymer subunits [11, 21, 63]. C. glutamicum is a high-GC content organism categorized into Actinobacteria, in which utilizing CRISPR-Cas9 to generate engineered expression system results in toxicity in target cells. Hence, using CRISPR-Cas systems, which recognize AT-rich canonical PAM site, seems to be more valuable as this leads to less toxicity in the recipient cells. Recently, it has been revealed that FnCpf1 can be used as an efficient genome editing tool in C. glutamicum to introduce insertions, gene deletions, and nucleotide exchanges [24]. The efficiency of presenting small changes made by CRISPR-Cpf1 joined together with single-strand DNA (ssDNA) donor template is estimated to be 86–100%. As an example, L-proline inhibition was successfully eliminated in C. glutamicum through codon saturation mutagenesis (modification from a wild-type amino acid to all other amino acids) at G149 of γ-glutamyl kinase using the CRISPR-Cpf1 system combined with ssDNA recombineering. Furthermore, it was indicated that N rounds of iterative Cpf1 genome editing are quicker (3N+4 or 3N+2 days) than traditional allelic exchange protocols (8N days) [24, 48]. Altogether, CRISPR-Cpf1 is a useful genome editing approach in bacteria such as, Corynebacterium, which are intolerant of being engineered by the CRISPR-Cas9 system. Cyanobacteria can convert CO2 into desired end product by using solar energy, which makes them an ideal organism for the production of bioproducts. However, using Cas9 to attain colony in each conjugation, encountered the protocol with Cas9 toxicity and was encountered with toxicity and low efficiency (<10 colonies). By utilizing Cpf1, which is non-toxic to Cyanobacteria, three models of cyanobacteria (Synechocystis, Anabaena, and Synechococcus), were engineered by inducing markerless knock-outs (KO), knock-ins (KI), and point mutations. This strategy is valuable because it will enable the researcher to distinguish various genes in an operon without forming polar effects of the foreign cassette [62]. Before it can be concluded that the CRISPR-Cpf1 system can be used widely spread as a genome editing tool in bacteria, it is necessary to be studied in further detailed experiments [68].
The efficiency of CRISPR-Cpf1 was also well proven in mammalian systems in two separate studies using Digenome-seq [30, 32] and GUIDE-seq analyses [31, 40, 60]. Kim et al. examined the efficiency of the four Cpf1 orthologs (LbCpf1, AsCpf1, FnCpf1, and MbCpf1) and in correlation with previous report [72], it was elucidated that LbCpf1 and AsCpf1 are the most efficient endonucleases in eukaryotic cells in combination with orthogonal and non-orthogonal crRNAs, albeit recently it was demonstrated that FnCpf1 can be employed in human cells as well [61]. Likewise, it was indicated that Cpf1 orthologs cannot tolerate mismatch at the 5′ PAM-proximal part while single or double mismatches at the 3′ PAM-distal part can be tolerated [31]. The frequency of target mutation for the Cpf1 endonuclease was estimated to be the same as Staphylococcus aureus Cas9 (SaCas9), 20 ± 5% and 19 ± 6% for AsCpf1 and LbCpf1, respectively [52] and lower mutation frequency compared with SpCas9 (32 ± 4%), at the same chromosomal target sites. To diminish the off-target effect index (OTI) for Cpf1, two approaches have been proposed: first, transfection of Cpf1 ribonucleoprotein (RNP) because the half-life of protein and RNA is shorter than DNA [35] and in this line, utilizing Cpf1 RNP successfully eliminated any concern about the off-target effects that had been induced when the plasmid was used as a cargo (OTI <0.0004). Second, using truncated crRNA [16] at 3′ region which decreased the frequency of indels at off-target sites up to ninefold, albeit the efficiency of using truncated crRNA for Cpf1 is limited owing to fact that most off-target sites have mismatches at 3′ PAM-distal region. By utilizing Digenome-seq, in vitro off-target digestion sites were estimated to be about 12 ± 5 and 6 ± 3 for AsCpf1 and LbCpf1, respectively, indicating high specificity of Cpf1 in eukaryotic cells compared to SpCas9 with 90 ± 30 digestion events [30, 31]. Kleinstiver et al. also verified that LbCpf1 and AsCpf1 are the most efficient Cpf1 orthologs in eukaryotic cells. Their results showed in concordance with an earlier report [31] that Cpf1 can tolerate single-base mismatch in 4–6 bases at 3′ PAM-distal region. Besides, it was discovered that 17–19 bases (from 23-nt crRNA) 5′ PAM-proximal region are important for robust Cpf1 activity and that deletion or extension of 4–6 bases at the 3′ end of the crRNA does not modify the efficiency of Cpf1 orthologs [40]. Also, the use of GUIDE-seq and deep sequencing analyses confirmed that Cpf1 endonuclease is highly specific in human cells in comparison with high-fidelity SpCas9 alternatives [39, 53]. It was also revealed that AsCpf1 DNase activity shows greater off-target effect, whereas its PAM-binding domain is more discriminating than LbCpf1. In contrast, the scaffold-binding region of LbCpf1 is more selective than that of AsCpf1, meaning that LbCpf1 can only act with its related scaffold [30, 40, 72]. In this line, generating Cpf1 variants with improved fidelity to reduce any trace of undetected off-target mutations is necessary to further improve the genome editing toolbox.
Cpf1 also represented its promising achievement in in vivo studies. Microinjecting the mRNA of Cpf1 orthologs, AsCpf1 and LbCpf1, and the corresponding crRNAs into mouse embryos to target Trp53 and Prkdc led to newborns harboring 70–80% mutation frequencies although the percentage of mutations at the Prkdc locus for AsCpf1 was estimated to be 18.2%. The off-target mutations with the frequency of 16.3% for LbCpf1 and 18.6% for AsCpf1 were recognized only at the sites with one base pair mismatch [37]. Furthermore, Electroporation of recombinant AsCpf1 RNP into one-cell embryos of mouse to target the Foxn1 locus resulted in 64% mutant without any detectable off-target mutation at the potential loci. Based on this achievement, electroporation is a powerful approach to create mouse models by using Cpf1 RNP [23]. To obtain a guide for designing appropriate gRNAs at different loci in human cells, an in vivo comprehensive study was recently carried out to evaluate the Cpf1 DNA cleavage activity at >11,000 target sequences. The gRNAs were delivered by lentiviral vectors into human cells to make cell library. After delivering Cpf1 by plasmid or lentiviral vector, the frequency of indels at the integrated synthetic target sequences was estimated by employing deep sequencing. As a result, an in silico web-tool has been established that is able to predict the indel frequency at on-target sites for AsCpf1 endonuclease (http://big.hanyang.ac.kr/cindel) [34]. The introduced high-throughput evaluation system promotes designing the well-organized and explicit gRNA for various target sites in human cells. A list of putative user-friendly Cpf1 gRNA designing software is presented in Table 1. Staggered cleavage at target sites by Cpf1 orthologs makes them an excellent candidate for inducing the homology-directed repair (HDR) pathway in host cells. It was indicated that Cpf1 orthologs, AsCpf1 and LbCpf1, are more efficient than SpCas9 orthologs (N. meningitidis, NmCas9; Streptococcus thermophilus, StCas9; and SaCas9) at inducing HDR in N2a mouse neuroblastoma cells. The average HDR inductions were estimated to be about 24% for LbCpf1 and 15% for AsCpf1 versus 13% for SaCas9, 9% for StCas9, and 3% for NmCas9 [59]. Altogether, these observations have introduced Cpf1 endonucleases as an effective genome editing tool besides the promiscuous SpCas9.
The RNase activity of Cpf1 provides the opportunity to process several mature crRNAs from a single pre-transcript, which is expressed under only one Pol III promoter, and consequently helps us to target several genomic sites simultaneously. However, Cas9 nuclease requires large constructs, or delivery of multiple expression plasmids to be able to perform multiplex genome editing. Multiplex genome editing for four different genes (EMX1, VEGFA, DNMT1, and GRIN2b) in HEK293T cells was successfully carried out when the CRISPR array was constructed in the order: 19-nt direct repeat with 23-nt guide. Likewise, multiplex targeting of neural genes (Mecp2, Nlgn3, and Drd1) was accomplished by using AsCpf1 in mouse primary cortical neurons and indel formation frequencies were estimated to be ~23% (Mecp2), ~38% (Nlgn3), and ~51% (Drd1) [73]. Using RNA processing activity of AsCpf1, delivery of several gRNAs for multiplex genome editing will be simplified in mammalian cells. Furthermore, the RNA processing activity of Cpf1 orthologs (AsCpf1 and LbCpf1) was also demonstrated for multiplex genome editing in mammalian cells, in which the CRISPR array was expressed using RNA polymerase II promoter. It was indicated that by increasing the length of crRNA array under a Pol III promoter, the expression of transcript decreases, but this reduced expression was not observed when the Pol II promoter was employed. Utilizing the Pol II-derived transcript for multiplex genome editing was even more efficient than the Pol III-derived transcript, perhaps because the Pol II-derived transcripts are able to be exported into the cytoplasm, where the transcripts might simply act with translated Cpf1. Taken together, Pol II- or Pol III-derived transcripts enable multiplex in vivo genome editing with viral vectors that can shelter a single promoter for the expression of multiple crRNAs and a promoter for the expression of Cpf1 endonuclease [79].
Repurposing CRISPR-Cas systems to regulate gene expression was first explained in Escherichia coli by using a catalytically dead-Cas9 protein, which is bound to target site without introducing DSB (CRISPR interference, CRISPRi) [49]. CRISPR-Cpf1 system can also be engineered to be a catalytically dead protein and used as a gene expression regulator in human cells. By introducing mutation at conserved residue, D880A, in Eubacterium eligens (EeCpf1) a novel tunable CRISPRi system was presented: DNA nuclease-deactivated EeCpf1 (EeddCpf1). This mutation completely disrupts the DNA cleavage activity but not the RNA processing activity of EeCpf1, which provides the opportunity to perform multiplex gene expression regulations. Furthermore, it has been found that EeddCpf1 has extensive gene repression (86.7%) when it is targeted to the complementary strand at the 5′-UTR or coding regions. This result does not concur with dCas9, but when EeddCpf1 was targeted to the promoter, no bias strand was observed [36]. These findings introduced the CRISPR-EeddCpf1 system as a gene expression regulator toolbox. AsCpf1 can also be converted to a DNA nuclease-deactivated AsCpf1 (AsddCpf1) by introducing E993A mutation in its RuvC-like domain to be used as a gene expression regulator as well. Similar to the EeddCpf1, AsddCpf1 showed bias strand in targeting the gene sequence but not when it was targeted to the promoter. Moreover, AsddCpf1 was highly efficient in multiplex gene repression in HEK293T cell line when simultaneously applied with a CRISPR array, which was ordered as previously mentioned [73, 74]. Fusing the transcriptional repressor or activator might enhance the efficiency of gene expression regulation similar to the fusion of ddCpf1 and three copies of the SRDX transcriptional repressor, which was performed in a plant to target the promoter region of miR159b (a non-coding RNA) [58].
Moreover, Cpf1 is efficient at correcting genetic mutation in human cells and mouse model. AsCpf1 and LbCpf1 were successfully used to correct Duchene muscular dystrophy (DMD) in human cardiomyocytes by either editing a nonsense mutation or reframing an out-of-frame deletion mutation through exon skipping. The reframing strategy was implemented by introducing inactivating mutation to splice acceptor of exon 51, which resulted in the skipping of out-of-frame deletion mutation to restore the open reading frame. Furthermore, utilizing Cpf1 orthologs, AsCpf1 and LbCpf1, indicated that DMD correction can be performed in mdx mice by targeting a nonsense mutation at exon 23 of the dystrophin gene and subsequently repairing the target site by HDR-mediated correction. LbCfp1 showed higher efficiency in mdx mice by yielding greater occurrence of indel. Full restoration of the dystrophin protein was achieved in LbCpf1-treated mice that showed 50% genomic correction [77].
CRISPR-Cpf1 also showed a promising result in editing driver mutations such as BRAF-V600E (1799T>A), which is a frequent mutation in multiple types of cancers. Disruption of mutant allele in patients can be considered as an efficient gene therapy approach. It was elucidated that only CRISPR-Cpf1 systems are able to selectively inactivate the mutant BRAF allele, while CRISPR-Cas9 system cannot discriminate mutant and normal forms and disrupt both alleles. In addition, CRISPR-Cas9 EQR variant (NGAG as PAM sequence) had no obvious gene editing events at the target sites. Based on these observations, the potential pertinence of Cpf1 in precise medicine through selective inactivation of gain-of-function mutations has been demonstrated [69].
CRISPR-Cpf1 as a groundbreaking genome editing tool in bacterial and mammalian cells has also imposed a revolution in plant genome engineering. Consistent with achievements in mammalian cells, studies have indicated that Cpf1 orthologs show little off-target effects, although off-target activities does not seem to be a serious concern in plant cells [33, 65, 70]. Recent advances in prokaryotic and eukaryotic genome engineering by using CRISPR-Cpf1 are summarized in Supplementary Table S1.
Conclusion
The CRISPR-Cpf1 system is highly promising as a genome editing tool. It is necessary to address the ambiguities regarding the biology of the type V (class II) CRISPR-Cas system, in particular; its spacer acquisition knowing that the CRISPR-Cpf1 system shares usual properties with class 1 systems. Mature crRNA biogenesis in the CRISPR-Cas9 system requires Cas9 protein, host RNase III, and tracrRNA, whereas the CRISPR-Cpf1 system seems to only need Cpf1 [13]. Elucidation of the RNA processing activity of Cpf1 offers the advantage of using multiplex genome editing in host cells. In line with this premise, multiplex genome editing approaches have recently been reported in mammalian and plant cells. In addition, it is extremely recommended to utilize this property of the CRISPR-Cpf1 system for multiplex targeting in industrial metabolite producers, such as C. glutamicum, where Cas9 results in lethal toxicity [24]. The PAM recognition feature of Cpf1 orthologs restricted to T-rich canonical form, 5′-TTTN-3′ or 5′-TTN-3′, but engineered Cpf1 variants (RR and RVR) has brought fascinating insights to expand the versatility and applicability of CRISPR-Cpf1 system in target hosts. The cleavage activity of CRISPR-Cpf1 system by creating RR and RVR variants is increased to every ~11 bp in coding sequences of human genome. Moreover, chemical modifications on crRNA (cr3′5F) and Cpf1 mRNA (full ψ-modification) have improved genome editing efficiency in mammalian cells. Taken together, engineered CRISPR-Cpf1 systems significantly maximized the genome editing efficiency without augmenting off-target effects. The promising results of Cpf1 orthologs in correcting human DMD mutation introduce CRISPR-Cpf1-mediated mutation correction as a powerful approach to remedy abnormalities associated with genetic disorders. Moreover, the following issues need to be addressed in future studies: the fusion of ddCpf1 protein to different mediators including transcriptional repressor or activator domains for efficient gene expression regulation, the fluorescent proteins for analyzing chromatin dynamics, the epigenetic modifier domains, and DNA-barcoding techniques for tracking cell lineages.
References
Alcon P, Montoya G, Stella S (2017) Assembly of Francisella novicida Cpf1 endonuclease in complex with guide RNA and target DNA. Acta Crystallogr F 73(Pt 7):409–415. https://doi.org/10.1107/S2053230X1700838X
Bae S, Park J, Kim JS (2014) Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30(10):1473–1475. https://doi.org/10.1093/bioinformatics/btu048
Bayat H, Omidi M, Rajabibazl M, Sabri S, Rahimpour A (2017) The CRISPR growth spurt: from bench to clinic on versatile small RNAs. J Microbiol Biotechnol 27(2):207–218. https://doi.org/10.4014/jmb.1607.07005
Charpentier E, Richter H, van der Oost J, White MF (2015) Biogenesis pathways of RNA guides in archaeal and bacterial CRISPR-Cas adaptive immunity. FEMS Microbiol Rev 39(3):428–441. https://doi.org/10.1093/femsre/fuv023
Chira S, Gulei D, Hajitou A, Zimta AA, Cordelier P, Berindan-Neagoe I (2017) CRISPR/Cas9: transcending the reality of genome editing. Mol Ther Nucleic Acids 7:211–222. https://doi.org/10.1016/j.omtn.2017.04.001
Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E (2011) CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature 471(7340):602–607. https://doi.org/10.1038/nature09886
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, Virgin HW, Listgarten J, Root DE (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. https://doi.org/10.1038/nbt.3437
Dong D, Ren K, Qiu X, Zheng J, Guo M, Guan X, Liu H, Li N, Zhang B, Yang D, Ma C, Wang S, Wu D, Ma Y, Fan S, Wang J, Gao N, Huang Z (2016) The crystal structure of Cpf1 in complex with CRISPR RNA. Nature 532(7600):522–526. https://doi.org/10.1038/nature17944
Doudna JA, Charpentier E (2014) Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 346(6213):1258096. https://doi.org/10.1126/science.1258096
Dow LE (2015) Modeling disease in vivo with CRISPR/Cas9. Trends Mol Med 21(10):609–621. https://doi.org/10.1016/j.molmed.2015.07.006
Eggeling L, Bott M (2015) A giant market and a powerful metabolism: L-lysine provided by Corynebacterium glutamicum. Appl Microbiol Biotechnol 99(8):3387–3394. https://doi.org/10.1007/s00253-015-6508-2
Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM (2013) Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods 10(11):1116–1121. https://doi.org/10.1038/nmeth.2681
Fagerlund RD, Staals RH, Fineran PC (2015) The Cpf1 CRISPR-Cas protein expands genome-editing tools. Genome Biol 16:251. https://doi.org/10.1186/s13059-015-0824-9
Fellmann C, Gowen BG, Lin PC, Doudna JA, Corn JE (2017) Cornerstones of CRISPR-Cas in drug discovery and therapy. Nat Rev Drug Discov 16(2):89–100. https://doi.org/10.1038/nrd.2016.238
Fonfara I, Richter H, Bratovic M, Le Rhun A, Charpentier E (2016) The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature. https://doi.org/10.1038/nature17945
Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK (2014) Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol 32:279–284. https://doi.org/10.1038/nbt.2808
Gao L, Cox DBT, Yan WX, Manteiga JC, Schneider MW, Yamano T, Nishimasu H, Nureki O, Crosetto N, Zhang F (2017) Engineered Cpf1 variants with altered PAM specificities. Nat Biotechnol. https://doi.org/10.1038/nbt.3900
Gao P, Yang H, Rajashankar KR, Huang Z, Patel DJ (2016) Type V CRISPR-Cas Cpf1 endonuclease employs a unique mechanism for crRNA-mediated target DNA recognition. Cell Res 26(8):901–913. https://doi.org/10.1038/cr.2016.88
Haeussler M, Schonig K, Eckert H, Eschstruth A, Mianne J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, Joly JS, Concordet JP (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17(1):148. https://doi.org/10.1186/s13059-016-1012-2
Haurwitz RE, Jinek M, Wiedenheft B, Zhou K, Doudna JA (2010) Sequence- and structure-specific RNA processing by a CRISPR endonuclease. Science 329(5997):1355–1358. https://doi.org/10.1126/science.1192272
Heider SA, Wendisch VF (2015) Engineering microbial cell factories: metabolic engineering of Corynebacterium glutamicum with a focus on non-natural products. Biotechnol J 10(8):1170–1184. https://doi.org/10.1002/biot.201400590
Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R (2008) Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. J Bacteriol 190(4):1401–1412. https://doi.org/10.1128/JB.01415-07
Hur JK, Kim K, Been KW, Baek G, Ye S, Hur JW, Ryu SM, Lee YS, Kim JS (2016) Targeted mutagenesis in mice by electroporation of Cpf1 ribonucleoproteins. Nat Biotechnol 34(8):807–808. https://doi.org/10.1038/nbt.3596
Jiang Y, Qian F, Yang J, Liu Y, Dong F, Xu C, Sun B, Chen B, Xu X, Li Y, Wang R, Yang S (2017) CRISPR-Cpf1 assisted genome editing of Corynebacterium glutamicum. Nat Commun 8:15179. https://doi.org/10.1038/ncomms15179
Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337(6096):816–821. https://doi.org/10.1126/science.1225829
Jinek M, Jiang F, Taylor DW, Sternberg SH, Kaya E, Ma E, Anders C, Hauer M, Zhou K, Lin S, Kaplan M, Iavarone AT, Charpentier E, Nogales E, Doudna JA (2014) Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343(6176):1247997. https://doi.org/10.1126/science.1247997
Karvelis T, Gasiunas G, Young J, Bigelyte G, Silanskas A, Cigan M, Siksnys V (2015) Rapid characterization of CRISPR-Cas9 protospacer adjacent motif sequence elements. Genome Biol 16:253. https://doi.org/10.1186/s13059-015-0818-7
Kato T, Takada S (2017) In vivo and in vitro disease modeling with CRISPR/Cas9. Brief Funct Genomics 16(1):13–24. https://doi.org/10.1093/bfgp/elw031
Kennedy EM, Cullen BR (2015) Bacterial CRISPR/Cas DNA endonucleases: a revolutionary technology that could dramatically impact viral research and treatment. Virology 479–480:213–220. https://doi.org/10.1016/j.virol.2015.02.024
Kim D, Kim S, Kim S, Park J, Kim JS (2016) Genome-wide target specificities of CRISPR-Cas9 nucleases revealed by multiplex Digenome-sEq. Genome Res 26(3):406–415. https://doi.org/10.1101/gr.199588.115
Kim D, Kim J, Hur JK, Been KW, Yoon SH, Kim JS (2016) Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat Biotechnol 34(8):863–868. https://doi.org/10.1038/nbt.3609
Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS (2015) Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nat Methods 12(3):237–243. https://doi.org/10.1038/nmeth.3284 (231 p following 243)
Kim H, Kim ST, Ryu J, Kang BC, Kim JS, Kim SG (2017) CRISPR/Cpf1-mediated DNA-free plant genome editing. Nat Commun 8:14406. https://doi.org/10.1038/ncomms14406
Kim HK, Song M, Lee J, Menon AV, Jung S, Kang YM, Choi JW, Woo E, Koh HC, Nam JW, Kim H (2017) In vivo high-throughput profiling of CRISPR-Cpf1 activity. Nat Methods 14(2):153–159. https://doi.org/10.1038/nmeth.4104
Kim K, Park SW, Kim JH, Lee SH, Kim D, Koo T, Kim KE, Kim JH, Kim JS (2017) Genome surgery using Cas9 ribonucleoproteins for the treatment of age-related macular degeneration. Genome Res 27(3):419–426. https://doi.org/10.1101/gr.219089.116
Kim SK, Kim H, Ahn WC, Park KH, Woo EJ, Lee DH, Lee SG (2017) Efficient transcriptional gene repression by type V-A CRISPR-Cpf1 from Eubacterium eligens. ACS Synth Biol. https://doi.org/10.1021/acssynbio.6b00368
Kim Y, Cheong SA, Lee JG, Lee SW, Lee MS, Baek IJ, Sung YH (2016) Generation of knockout mice by Cpf1-mediated gene targeting. Nat Biotechnol 34(8):808–810. https://doi.org/10.1038/nbt.3614
Kleinstiver B, Prew M, Tsai S, Topkar V, Nguyen N, Zheng Z (2015) Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature 523:4
Kleinstiver BP, Pattanayak V, Prew MS, Nguyen QS, Zheng NT, Joung Z JK (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. https://doi.org/10.1038/nature16526
Kleinstiver BP, Tsai SQ, Prew MS, Nguyen NT, Welch MM, Lopez JM, McCaw ZR, Aryee MJ, Joung JK (2016) Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat Biotechnol 34(8):869–874. https://doi.org/10.1038/nbt.3620
Komor AC, Badran AH, Liu DR (2017) CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell 168(1–2):20–36. https://doi.org/10.1016/j.cell.2016.10.044
Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E (2016) CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44(W1):W272–W276. https://doi.org/10.1093/nar/gkw398
Lei C, Li SY, Liu JK, Zheng X, Zhao GP, Wang J (2017) The CCTL (Cpf1-assisted Cutting and Taq DNA ligase-assisted Ligation) method for efficient editing of large DNA constructs in vitro. Nucleic Acids Res 45(9):e74. https://doi.org/10.1093/nar/gkx018
Li B, Zhao W, Luo X, Zhang X, Li C, Zeng C, Dong Y (2017) Engineering CRISPR-Cpf1 crRNAs and mRNAs to maximize genome editing efficiency. Nat Biomed Eng. https://doi.org/10.1038/s41551-017-0066
Mohanraju P, Makarova KS, Zetsche B, Zhang F, Koonin EV, van der Oost J (2016) Diverse evolutionary roots and mechanistic variations of the CRISPR-Cas systems. Science 353(6299):aad5147. https://doi.org/10.1126/science.aad5147
Nam KH, Haitjema C, Liu X, Ding F, Wang H, DeLisa MP, Ke A (2012) Cas5d protein processes pre-crRNA and assembles into a cascade-like interference complex in subtype I-C/Dvulg CRISPR-Cas system. Structure 20(9):1574–1584. https://doi.org/10.1016/j.str.2012.06.016
Nishimasu H, Yamano T, Gao L, Zhang F, Ishitani R, Nureki O (2017) Structural basis for the altered PAM recognition by engineered CRISPR-Cpf1. Mol Cell. https://doi.org/10.1016/j.molcel.2017.04.019
Okibe N, Suzuki N, Inui M, Yukawa H (2011) Efficient markerless gene replacement in Corynebacterium glutamicum using a new temperature-sensitive plasmid. J Microbiol Methods 85(2):155–163. https://doi.org/10.1016/j.mimet.2011.02.012
Qi L, Larson M, Gilbert L, Doudna J, Weissman J, Arkin A, Lim W (2013) Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152(5):10. https://doi.org/10.1016/j.cell.2013.02.022
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8(11):28. https://doi.org/10.1038/nprot.2013.143
Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, Zhang F (2013) Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154(6):1380–1389. https://doi.org/10.1016/j.cell.2013.08.021
Ran FA, Cong L, Yan WX, Scott DA, Gootenberg JS, Kriz AJ, Zetsche B, Shalem O, Wu X, Makarova KS, Koonin EV, Sharp PA, Zhang F (2015) In vivo genome editing using Staphylococcus aureus Cas9. Nature 520(7546):186–191. https://doi.org/10.1038/nature14299
Slaymaker IM, Gao L, Zetsche B, Scott DA, Yan WX, Zhang F (2016) Rationally engineered Cas9 nucleases with improved specificity. Science 351(6268):84–88. https://doi.org/10.1126/science.aad5227
Stella S, Alcon P, Montoya G (2017) Structure of the Cpf1 endonuclease R-loop complex after target DNA cleavage. Nature 546(7659):559–563. https://doi.org/10.1038/nature22398
Stemmer M, Thumberger T, Del Sol Keyer M, Wittbrodt J, Mateo JL (2015) CCTop: an intuitive, flexible and reliable CRISPR/Cas9 target prediction tool. PLoS ONE 10(4):e0124633. https://doi.org/10.1371/journal.pone.0124633
Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA (2014) DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507(7490):62–67. https://doi.org/10.1038/nature13011
Szczelkun MD, Tikhomirova MS, Sinkunas T, Gasiunas G, Karvelis T, Pschera P, Siksnys V, Seidel R (2014) Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc Natl Acad Sci USA 111(27):9798–9803. https://doi.org/10.1073/pnas.1402597111
Tang X, Lowder LG, Zhang T, Malzahn AA, Zheng X, Voytas DF, Zhong Z, Chen Y, Ren Q, Li Q, Kirkland ER, Zhang Y, Qi Y (2017) A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat Plants 3:17018. https://doi.org/10.1038/nplants.2017.18
Toth E, Weinhardt N, Bencsura P, Huszar K, Kulcsar PI, Talas A, Fodor E, Welker E (2016) Cpf1 nucleases demonstrate robust activity to induce DNA modification by exploiting homology directed repair pathways in mammalian cells. Biol Direct 11:46. https://doi.org/10.1186/s13062-016-0147-0
Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, Aryee MJ, Joung JK (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat Biotechnol 33(2):187–197. https://doi.org/10.1038/nbt.3117
Tu M, Lin L, Cheng Y, He X, Sun H, Xie H, Fu J, Liu C, Li J, Chen D, Xi H, Xue D, Liu Q, Zhao J, Gao C, Song Z, Qu J, Gu F (2017) A ‘new lease of life’: FnCpf1 possesses DNA cleavage activity for genome editing in human cells. Nucleic Acids Res. https://doi.org/10.1093/nar/gkx783
Ungerer J, Pakrasi HB (2016) Cpf1 is a versatile tool for CRISPR genome editing across diverse species of cyanobacteria. Sci Rep 6:39681. https://doi.org/10.1038/srep39681
Wendisch VF, Jorge JMP, Perez-Garcia F, Sgobba E (2016) Updates on industrial production of amino acids using Corynebacterium glutamicum. World J Microbiol Biotechnol 32(6):105. https://doi.org/10.1007/s11274-016-2060-1
White MF (2016) Cpf1 shape-shifts for streamlined CRISPR cleavage. Nat Struct Mol Biol 23(5):365–366. https://doi.org/10.1038/nsmb.3225
Xu R, Qin R, Li H, Li D, Li L, Wei P, Yang J (2017) Generation of targeted mutant rice using a CRISPR-Cpf1 system. Plant Biotechnol J 15(6):713–717. https://doi.org/10.1111/pbi.12669
Yamano T, Zetsche B, Ishitani R, Zhang F, Nishimasu H, Nureki O (2017) Structural basis for the canonical and non-canonical PAM recognition by CRISPR-Cpf1. Mol Cell 67(4):633–645 e633. https://doi.org/10.1016/j.molcel.2017.06.035
Yamano T, Nishimasu H, Zetsche B, Hirano H, Slaymaker IM, Li Y, Fedorova I, Nakane T, Makarova KS, Koonin EV, Ishitani R, Zhang F, Nureki O (2016) Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell 165(4):949–962. https://doi.org/10.1016/j.cell.2016.04.003
Yan MY, Yan HQ, Ren GX, Zhao JP, Guo XP, Sun YC (2017) CRISPR-Cas12a-assisted recombineering in bacteria. Appl Environ Microbiol 83 (17). https://doi.org/10.1128/AEM.00947-17
Yang M, Wei H, Wang Y, Deng J, Tang Y, Zhou L, Guo G, Tong A (2017) Targeted disruption of V600E-mutant BRAF gene by CRISPR-Cpf1. Mol Ther Nucleic Acids 8:450–458. https://doi.org/10.1016/j.omtn.2017.05.009
Zaidi SS, Mahfouz MM, Mansoor S (2017) CRISPR-Cpf1: a new tool for plant genome editing. Trends Plant Sci 22(7):550–553. https://doi.org/10.1016/j.tplants.2017.05.001
Zetsche B, Strecker J, Abudayyeh OO, Gootenberg JS, Scott DA, Zhang F (2017) A survey of genome editing activity for 16 Cpf1 orthologs. bioRxiv. https://doi.org/10.1101/134015
Zetsche B, Gootenberg JS, Abudayyeh OO, Slaymaker IM, Makarova KS, Essletzbichler P, Volz SE, Joung J, van der Oost J, Regev A, Koonin EV, Zhang F (2015) Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 163(3):759–771. https://doi.org/10.1016/j.cell.2015.09.038
Zetsche B, Heidenreich M, Mohanraju P, Fedorova I, Kneppers J, DeGennaro EM, Winblad N, Choudhury SR, Abudayyeh OO, Gootenberg JS, Wu WY, Scott DA, Severinov K, van der Oost J, Zhang F (2017) Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat Biotechnol 35(1):31–34. https://doi.org/10.1038/nbt.3737
Zhang X, Wang J, Cheng Q, Zheng X, Zhao G, Wang J (2017) Multiplex gene regulation by CRISPR-ddCpf1. Cell Discov 3:17018. https://doi.org/10.1038/celldisc.2017.18
Zhang Y, Rajan R, Seifert HS, Mondragon A, Sontheimer EJ (2015) DNase H activity of Neisseria meningitidis Cas9. Mol Cell 60(2):242–255. https://doi.org/10.1016/j.molcel.2015.09.020
Zhang Y, Heidrich N, Ampattu BJ, Gunderson CW, Seifert HS, Schoen C, Vogel J, Sontheimer EJ (2013) Processing-independent CRISPR RNAs limit natural transformation in Neisseria meningitidis. Mol Cell 50(4):488–503. https://doi.org/10.1016/j.molcel.2013.05.001
Zhang Y, Long C, Li H, McAnally JR, Baskin KK, Shelton JM, Bassel-Duby R, Olson EN (2017) CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv 3(4):e1602814. https://doi.org/10.1126/sciadv.1602814
Zhang Y, Ge X, Yang F, Zhang L, Zheng J, Tan X, Jin ZB, Qu J, Gu F (2014) Comparison of non-canonical PAMs for CRISPR/Cas9-mediated DNA cleavage in human cells. Sci Rep 4:5405. https://doi.org/10.1038/srep05405
Zhong G, Wang H, Li Y, Tran MH, Farzan M (2017) Cpf1 proteins excise CRISPR RNAs from mRNA transcripts in mammalian cells. Nat Chem Biol. https://doi.org/10.1038/nchembio.2410
Acknowledgements
The authors wish to thank Medical Nano-Technology & Tissue Engineering Research Center and School of Advanced Technologies in Medicine, Shahid Beheshti University of Medical Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bayat, H., Modarressi, M.H. & Rahimpour, A. The Conspicuity of CRISPR-Cpf1 System as a Significant Breakthrough in Genome Editing. Curr Microbiol 75, 107–115 (2018). https://doi.org/10.1007/s00284-017-1406-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-017-1406-8