
Abstract
Recombineering is a homologous-based DNA cloning and modification technique. Recombineering-mediated chromosomal gene knock-in usually involves a selectable/counterselectable cassette. Though a variety of selectable/counterselectable cassettes were developed; however, a specifically designed gene deletion strain or minimal medium is often required. Herein, we describe a novel selectable/counterselectable cassette Plac-ccdB-aacC1 in which aacC1 (gentamicin resistance gene) is used as the selectable marker for the homologous arm-flanked cassette knock-in, while the IPTG inducible ccdB gene is used as the counterselectable marker for chromosomal gene knock-in. The counterselection is achieved via supplementing 1 mM IPTG in the LB agar medium. An oligonucleotide designed to evade the mismatch repair system was utilized to engineer an Escherichia coli DH10B-derived gyrA462 strain that was used to as the host for the plasmid harboring the Plac-ccdB-aacC1 cassette. By using the Plac-ccdB-aacC1 cassette, a linear–linear homologous recombination (LLHR) system was generated by knocking a 6.2 kb araC-PBAD-redγ-recET-recA DNA fragment into the E. coli DH10B chromosome. The functional of the LLHR recombineering system was characterized by cloning of the target DNA from PCR product as well as from the genomic DNA mixture. The Plac-ccdB-aacC1 cassette will be a useful tool in E. coli recombineering.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Recombineering is a homologous recombination-based technology that is widely used for bacterial genome editing, episomal DNA cloning, and modification [9, 10]. Selectable/counterselectable cassette is often used for recombineering-mediated Escherichia coli chromosomal knock-in. A variety of counterselectable markers were developed, including sacB [17], galK [15], thyA [16], tolC [4], rpsL [14], and tetA-sacB [8]. However, use of these counterselectable markers often involves a specific gene deletion strain which involves more steps or minimal medium which retards the strain’s growth. Other shortcoming, such as accumulation of mutations in E. coli even when grown without sucrose in using sacB, is also worthy consideration.
Toxic gene ccdB in the toxin–antitoxin system [1] was recently developed as an efficient counterselectable marker [13]; yet the delicate, two plasmid-based toxin–antitoxin expression system complicated the screening.
To improve the process, herein a Plac-ccdB-aacC1 cassette in which ccdB was driven by IPTG (isopropyl-β-d-1-thiogalactopyranoside) inducible Plac promoter was developed as a selectable/counterselectable marker. In the cassette, the aacC1 (gentamicin resistance gene) was used for the first step recombinase-catalyzed homologous arm (HA) flanked Plac-ccdB-aacC1 cassette knock-in, then the cassette was replaced by the same HA-flanked target gene though the second recombineering step via the counterselection by the agar medium with IPTG. Functional characterization of the selectable/counterselectable system was demonstrated by knocking a 6.2 kb fragment (araC-PBAD-redγ-recET-recA) into the E. coli DH10B chromosome, generating a linear–linear homologous recombination (LLHR) recombineering system. The LLHR system was characterized by cloning of a 0.8 kb chloramphenicol resistance gene (cat) from linear PCR product as well as a 3.2 kb lacZ gene from E. coli MG1655 genomic DNA. Though oligonucleotide-mediated recombineering, a highly transformative E. coli strain was also engineered to host the Plac-ccdB-aacC1 cassette template plasmid. The cassette has the potential to simplify the E. coli chromosomal gene knock-in as well as other recombineering applications.
Materials and Methods
Bacterial Strains, Culture, and Plasmids
Escherichia coli cells were grown in Luria–Bertani (LB) broth supplemented, when appropriate, with the following antibiotics: Ampicillin sodium (Ap, 50 μg/ml), Kanamycin sulfate (Km, 30 μg/ml), or Gentamicin sulfate (Gm, 25 μg/ml). E. coli DH10B was used as general cloning host; E. coli LS027 generated in this study was used as the cloning host of ccdB gene harboring plasmids. A final concentration of 25 μg/ml heat-inactivated cTc (chlortetracycline hydrochloride) was used for homing endonuclease I-SceI induction. Restriction enzymes were products of NEB, USA. Gel extraction kit was purchased from Qiagen, Germany. The strains and plasmids used in this study are listed in Table S1; oligonucleotides used in this study are listed in Table S2. Details of the vector construction are provided in Supplementary Material.
Molecular Biology and Recombineering Manipulations
Escherichia coli electrocompetent cells preparation, DNA transformation, plasmid extraction, restriction enzyme digestion, PCR, and E. coli MG1655 genomic DNA preparation with lysozyme treatment and phenol–chloroform extraction were carried out with standard procedures. Recombineering manipulation was carried out as described [3].
Results and Discussion
Construction of a Highly Transformative ccdB Host Strain
A highly transformative E. coli strain for harboring ccdB gene was firstly constructed by introducing a gyrA462 mutation (arginine to cysteine mutation at the GyrA amino acid 462 position) via oligonucleotide-mediated recombineering. Co-transformation of 5 pmol of GYRA2 designed to evade mismatch repair by adding four mutations at wobble positions [11] and 100 ng of ccdB-harboring, kanamycin resistance plasmid pMK2010 [6] into the Red-competent cells of E. coli DH10β F’DOT [7] containing Red helper plasmid pLS3021 resulted in 65 KmR clones. Genotype analysis revealed that seven of ten clones exhibited expected mutations. pMK2010 was eliminated from the engineered strain by transforming of a same pUC replicon-based, Ap resistance, self-cleavage plasmid pLS3050; pLS3050 was subsequently removed by culturing the cells with cTc induction. Three of 45 clones tested were ApS; one strain was named as E. coli LS027. Compared with that of DB3.1, E. coli LS027 exhibits approximately 100-fold of transformation efficiency (2.1 × 108 transformants/μg vs 3.2 × 106 transformants/μg) using pKD4 [4] as plasmid DNA. The strain construction scheme and genotype sequencing results are illustrated in Fig. 1.

Scheme shown a gyrA462 mutation via oligo-mediated recombineering. Ηomologous recombination between GYRA2 and its chromosomal allele resulted in expected mutations (a). The triplets of the target region of the gyrA gene are indicated for the wild-type (b) and gyrA462 mutant E. coli LS027 (c). Note that incorporation of GYRA2 changes a total change of 5 bases (red sequences that are indicated by arrows in (c) yielding a single amino acid change (R462C). Primers for screening (R483–R484) are indicated which generated a 341 bp amplicon (Color figure online)
Inducible Toxic Gene ccdB as a Novel Counterselectable Marker
A R6K replicon-based, selectable/counterselectable Plac-ccdB-aacC1 cassette template plasmid pLS3161 was constructed by fusing ccdB under the IPTG inducible Plac promoter with aacC1. The function was the 1165 bp cassette was characterized by construction of a chromosome-based LLHR recombineering system.
One microgram of 50 bp HA-flanked Plac-ccdB-aacC1 cassette amplified from pLS3161 was electroporated into the Red-induced electrocompetent cells of E. coli DH10B harboring pLS3021. The HAs were designed according to the truncated lacZ region of E. coli DH10B genome. A total of 218 clones were obtained under gentamicin selection; the clones showed no visible growth retardation, implying that uninduced CcdB exerts no observable deleterious effect. By contrast, 48 of 50 clones showed no growth when streaked on LB agar plate supplemented with 1 mM IPTG indicating the high toxicity of induced CcdB. The resulting strain, E. coli LS3125, was Red-induced and electroporated with 1 μg of targeting DNA released from pLS3185. Sixty-four IPTG resistant clones were generated, and eight out 10 clones analyzed were correct in which the Plac-ccdB-aacC1 cassette was replaced by the araC-PBAD-redγ-recET-recA fragment. After pLS3021 elimination through non-selective passages, a LLHR recombineering strain E. coli LS3130 was obtained. The scheme for the inducible ccdB counterselection-based strain construction and genotype analysis are shown in Fig. 2.

Schematic representation of ccdB counterselection for the construction of a LLHR recombineering system. a Homologous recombination between the HAs in the targeting DNA and its chromosomal allele generated E. coli LS3125. The second recombineering step generated a LLHR strain E. coli LS3130 by replacing the Plac-ccdB-aacC1 cassette by a 6.2 kb araC-PBAD-redγ-recET-recA DNA fragment under IPTG counterselection. b Genotype analysis. Lane 1 DNA marker, lane 2 and 3 are amplicons amplified from E. coli DH10B and E. coli LS3125 with primer set R2303–R2304, respectively, lane 4, 5, and 6 are amplicons amplified from E. coli LS3130 with primer sets R2303–R2317, R2319–R2320, and R2318–R2304, respectively. The sizes of the amplicons are indicated at (a)
Recombineering System Characterization
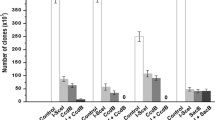
Cloning performance of E. coli LS3130 and its comparison with other recombineering systems were carried by cloning of cat gene and lacZ gene. For cat gene cloning, 100 ng of HA-flanked p15A-kan amplified from pLS3163 with primers R2321–R2322 and 1 μg of HA-flanked cat amplified from pBAD322C [2] with primers R2323–R2324 were co-electroporated into each recombineering system. For lacZ gene cloning, 1 μg of HA-flanked p15A-kan amplified from pLS3163 with primers R2327–R2328 and 5 μg of sheared E. coli MG1655 genomic DNA mixture were co-electroporated into each recombineering system. For every recombineering, ten clones were subjected to plasmid extraction, restriction enzymes digestions, and DNA sequencing. As shown in Table 1, the decreasing order of cloning efficiency is plasmid-based LLHR system pLS3028, chromosome-based LLHR system E. coli LS3130, plasmid-based linear–circular homologous recombination (LCHR) system pLS3021, and chromosome-based LCHR system LS-GR [12]. This result consists with the finding of Fu et al. [5] that LLHR shows higher cloning efficiency than LCHR in cloning of linear DNA molecules. Although E. coli LS3130 was ~10-fold less efficient than that of pLS3028, it circumvents the plasmid transformation and elimination steps and therefore is more convenient for DNA manipulation.
Inducible ccdB as a counterselectable marker circumvents the requirement of an antitoxin ccdA expression plasmid, making it a better choice for gene editing. Highly efficient counterselection can be realized by simply supplementing IPTG in the rich LB medium. The small size of the Plac-ccdB-aacC1 cassette is also easy for PCR amplification and genotyping. Besides E. coli LS3130 construction, we also knocked mkan fragment [16] and I-SceI gene into the chromosome of E. coli MG1665 via ccdB counterselection (data not shown). In summary, the inducible ccdB could be a powerful counterselectable marker for E. coli recombineering.
References
Bernard P, Couturier M (1992) Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J Mol Biol 226:735–745
Cronan JE (2006) A family of arabinose-inducible Escherichia coli expression vectors having pBR322 copy control. Plasmid 55:152–157
Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA 97:6640–6645
DeVito JA (2008) Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res 36:e4
Fu J, Bian X, Hu S, Wang H, Huang F, Seibert PM, Plaza A, Xia L, Muller R, Stewart AF, Zhang Y (2012) Full-length RecE enhances linear–linear homologous recombination and facilitates direct cloning for bioprospecting. Nat Biotechnol 30:440–446
House BL, Mortimer MW, Kahn ML (2004) New recombination methods for Sinorhizobium meliloti genetics. Appl Environ Microbiol 70:2806–2815
Li MZ, Elledge SJ (2005) MAGIC, an in vivo genetic method for the rapid construction of recombinant DNA molecules. Nat Genet 37:311–319
Li XT, Thomason LC, Sawitzke JA, Costantino N, Court DL (2013) Positive and negative selection using the tetA-sacB cassette: recombineering and P1 transduction in Escherichia coli. Nucleic Acids Res 41:e204
Murphy KC (1998) Use of bacteriophage lambda recombination functions to promote gene replacement in Escherichia coli. J Bacteriol 180:2063–2071
Murphy KC (2016) λ Recombination and recombineering. EcoSal Plus. doi:10.1128/ecosalplus.ESP-0011-2015
Sawitzke JA, Costantino N, Li XT, Thomason LC, Bubunenko M, Court C, Court DL (2011) Probing cellular processes with oligo-mediated recombination and using the knowledge gained to optimize recombineering. J Mol Biol 407:45–59
Song J, Dong H, Ma C, Zhao B, Shang G (2010) Construction and functional characterization of an integrative form λ Red recombineering Escherichia coli strain. FEMS Microbiol Lett 309:178–183
Wang H, Bian X, Xia L, Ding X, Muller R, Zhang Y, Fu J, Stewart AF (2014) Improved seamless mutagenesis by recombineering using ccdB for counterselection. Nucleic Acids Res 42:e37
Wang S, Zhao Y, Leiby M, Zhu J (2009) A new positive/negative selection scheme for precise BAC recombineering. Mol Biotechnol 42:110–116
Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG (2005) Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33:e36
Wong QN, Ng VC, Lin MC, Kung HF, Chan D, Huang JD (2005) Efficient and seamless DNA recombineering using a thymidylate synthase A selection system in Escherichia coli. Nucleic Acids Res 33:e59
Zhang Y, Buchholz F, Muyrers JP, Stewart AF (1998) A new logic for DNA engineering using recombination in Escherichia coli. Nat Genet 20:123–128
Acknowledgements
We than Dr. John Cronan, Dr. Stephen Ellege and Dr. Micheal Khan for kindly providing the strains and plasmids used in this research. Funding was provided by National Natural Science Foundation of China (81273412).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhang, Q., Yan, Z., Xu, Y. et al. Characterization of Inducible ccdB Gene as a Counterselectable Marker in Escherichia coli Recombineering. Curr Microbiol 74, 961–964 (2017). https://doi.org/10.1007/s00284-017-1273-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-017-1273-3