
Abstract
Francisella tularensis, the causative agent of tularemia, has attained the status of one of the high priority agents that could be used in the act of bioterrorism. Currently, there is no licensed vaccine for this highly infectious intracellular pathogen. Being a listed ‘Category A’ agent of the U.S. Center for Disease Control and Prevention (CDC), vaccines and therapeutics are immediately required against this pathogen. In this study, an immunoproteomic approach based on the techniques of 2-dimensional gel electrophoresis (2DE) and immunoblotting combined with mass spectrometry (MS) was used for elucidation of immunogenic components and putative vaccine candidates. Whole-cell soluble protein extract of F. tularensis LVS (Ft LVS) was separated by 2DE, and immunoblots were developed with sera raised in rabbit after immunization with heat-killed Ft LVS. A total of 28 immunoreactive proteins were identified by tandem mass spectrometry. Rabbit immunoproteome of F. tularensis was compared with those previously reported using sera from human patients and in murine model. Out of 28 immunoreactive proteins identified in this study, 12 and 17 overlapping proteins were recognized by human and murine sera, respectively. Nine proteins were found immunogenic in all the three hosts, while eight new immunogenic proteins were found in this study. Identified immunoreactive proteins may find application in design and development of protein subunit vaccine for tularemia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Francisella tularensis is a highly infectious zoonotic pathogen causing tularemia in humans. It is a non-spore forming, Gram-negative, aerobic intracellular bacterium with low infectious dose (<10 CFU) and high mortality rate, if left untreated. First identified in 1911 in ground squirrels in Tulare County of California, F. tularensis is comprised of four recognized subspecies: tularensis, holarctica, novicida, and mediasiatica. Out of these, subspecies tularensis is considered the most virulent in humans, while subspecies holarctica is the cause of most of the naturally occurring cases of tularemia [11].
Lower infective dose, ease of aerosolization, broad host range, and pulmonary route of infection qualifies F. tularensis as an agent of choice for bioterrorism [10]. Much of the research in recent years on F. tularensis is focused on the development of an effective vaccine against it. Hitherto, no licensed vaccine exists against this pathogen; although an attenuated live vaccine strain (LVS) has been used that gives partial protection to humans against respiratory challenge. Use of Ft LVS as vaccine is fraught with shortcomings due to the ambiguous source of attenuation and concern about reversion to a virulent phenotype [28].
Host immune response to Francisella infection and protection rendered by Ft LVS is poorly understood. Similar to other intracellular pathogens, protection is mainly due to T cell-based immune response against F. tularensis infection. However, harmony between humoral and cell-mediated immunity is essential to attain absolute protection against F. tularensis, and elevated antibody titer has been observed in both animals and humans following vaccination or exposure with F. tularensis [19]. Recently also, B cell functions have been shown to play important role in protecting BALB/cByJ mice against LVS lethal challenge vaccinated with suboptimal vaccines [7].
Immunoproteomics is a powerful method to explore immunogenic antigens as safe subunit vaccine to protect against pathogenic bacteria, and this approach has been used for a variety of microorganisms [9]. The immune response to LVS has been studied in depth in mice [3, 12, 15, 31–33], and the pattern of their susceptibility has been found different than humans. Previous studies have examined the immunogenicity in murine and human models [14, 17, 18], but the results have not been correlated using other animal models. Unlike humans, mice are highly responsive to all the subspecies of F. tularensis. Therefore, extrapolation of immunogenicity data from murine model to humans is not likely to be entirely predictive [22]. For that reason, successful development of any vaccine candidate against F. tularensis will require the use of other animal models to confirm efficacy and safety.
Rabbits constitute a valuable model for Francisella as they reflect the key patho-physiological features of the human disorder. In addition to showing gross pathology comparable to humans infected with virulent F. tularensis, rabbits are more resistant to Ft LVS [25]. Thus, further investigations will be useful to characterize the immune response of rabbits to F. tularensis to make predictive correlation of protection in humans. Therefore, this study focused on identifying immunogenic proteins of Francisella in a rabbit model with a view that these may be useful in developing a subunit vaccine for protection against tularemia.
Materials and Methods
Bacterial Cell Culture and Reagents
Francisella tularensis type B LVS (NCTC 10857), obtained from Health Protection Agency, UK was cultivated on cysteine heart agar (Difco, Germany) supplemented with 9% sheep blood at 37 °C for 48h in BSL-3 laboratory, DRDE, Gwalior. Briefly, a single colony was inoculated into Tryptic Soy Broth (Difco Laboratories) containing 0.1% cysteine and incubated overnight at 37 °C under continuous agitation. All chemicals and reagents were procured from Sigma-Aldrich, USA unless if not specified.
Preparation of Whole-Cell Extract
Bacteria were harvested by centrifugation at 10,000 g for 10 min, and the cell pellet was washed twice with ice-cold sterile distilled water. Cell pellet was resuspended in modified lysis buffer (7 M urea, 2 M thiourea, 1.0% NP-40, 4% CHAPS, and 1% DTT) as described earlier [31]. Centrifugation at 8000×g for 10 min at 4 °C resulted in total soluble protein in the supernatant fraction. The protein concentration of the bacterial lysate was quantified using the Bicinchoninic acid kit as per manufacturer’s instruction (Sigma, USA). One milligram of protein in the solution was precipitated with trichloroacetic acid (TCA) at a final concentration of 10% (w/v). After overnight incubation at 4 °C, the protein pellet was collected by centrifugation (10,000×g, 4 °C, and 10 min) and washed twice with acetone. The pellet was air-dried for 5 min and stored at −80 °C until use.
Serum Samples
Animal experiments were approved by the Institutional Animal Ethical Committee at DRDE, Gwalior, wide registration number 37/1999/CPCSEA; and Institutional Biosafety Committee (IBSC), wide protocol number ISBC/12/BT/DVK/4. Efforts were made to ensure minimum suffering of animals during experimentation. Heat-killed cells were prepared by incubating at 65 °C for 2 h on circulating water bath. Anti-Ft LVS serum was raised in New Zealand white rabbit by subcutaneous injection of heat-killed Ft LVS strain NCTC 10857 (1010 CFU). Blood was drawn from marginal ear vein of rabbit 42 days after immunization with Ft LVS, and serum was prepared by centrifugation (10,000×g, 4 °C, 10 min) and stored at -20 °C until use.
Two-Dimensional Gel Electrophoresis
The protein pellet was resuspended in modified sample rehydration buffer (7 M urea, 2 M thiourea, 1.0% NP-40, 4% CHAPS, 1% DTT, and 0.5% v/v IPG buffer pH 3–10) as described earlier [31]. For the first dimension, the extracted proteins were separated using immobilized pH gradient strips (IPG) of 7 cm size with a pH range from 5 to 8 (Bio-Rad, USA) using 500 μg protein in 150 μl rehydration solution after overnight passive rehydration. Proteins were focused at 10,000 Vh at 20 °C under mineral oil on a Protean IEF Cell (Bio-Rad, USA). Subsequently, proteins were reduced and alkylated by incubating the strips for 10 min in 2 ml equilibrium buffer I (6 M urea, 20% w/v glycerol, 2% w/v SDS, and 1% w/v DTT in 50 mM Tris/HCl buffer, pH 8.8) followed by another 10-min incubation in equilibrium buffer II (6 M urea, 20% w/v glycerol, 2% w/v SDS, and 4% w/v iodoacetamide in 375 mM Tris/HCl buffer, pH 8.8). After the equilibration steps, strips were transferred to 12% SDS–PAGE for the second-dimension electrophoresis. Second-dimension polyacrylamide gels were run in duplicate; first one was used for blotting and the second was stained with Coomassie Brilliant Blue R to serve as a 2D reference map. Gel images were captured by GS800 densitometer (Bio-Rad, USA).
Immunoblotting
Proteins separated by 2D PAGE were electroblotted onto PVDF membranes (Immobilon-P, 0.45 µm, Millipore) at 100 V for 1 h using Trans-Blot Cell (Bio-Rad, USA). Membranes were incubated overnight in PBS containing 5% w/v skimmed milk powder at 4 °C with constant shaking. Following three 10-min washes with PBST, the PVDF membrane was incubated with rabbit anti-Ft LVS serum diluted 1:8000 in PBST containing 2% w/v skimmed milk powder. Similarly, diluted healthy rabbit serum was used as control. After incubation for 1 h at room temperature with gentle shaking, blots were washed with PBST followed by incubation with peroxidase-conjugated goat anti-rabbit immunoglobulins (diluted 1:1000 in PBS) for 1 h at room temperature. Reactive spots were visualized using 3, 3′-diaminobenzidine (DAB) in the presence of hydrogen peroxide, and images were captured using GS800 densitometer (Bio-Rad, USA). Immunoblotting experiments were conducted in duplicate to ensure reproducibility.
In-Gel Digestion
In-gel digestion was carried out using the method previously described [20]. Briefly, gel pieces corresponding to immunoblots were excised from 2DE gels and destained at room temperature with 200 µl of 50 mM NH4HCO3 / acetonitrile (1:1, v/v) for 1 h. Proteins were reduced and alkylated and after drying the gel pieces with acetonitrile (ACN), 100 ng trypsin (in 50 mM NH4HCO3) was added to each piece. Tryptic digestion was carried out overnight at 37 °C. Peptides were extracted with 50% ACN and 0.1% trifluoroacetic acid (TFA), vacuum dried, and resuspended in 5 µl of 50% ACN and 0.01% TFA before MS analysis.
Identification of Protein Spots by Tandem Mass Spectrometry
Proteins, after tryptic digestion, were identified by MALDI TOF/TOF Analyzer (4800, AB Sciex, USA) as described earlier [20]. Digested peptides were mixed with 1:1 volume of the CHCA matrix solution (10 mg/ml in 50% ACN and 0.1% TFA) and spotted onto the target plate. Digested proteins were subjected to identification using conditions as previously described with some modifications [20]. Briefly, a six component peptide standard was used in a mass range of 905–3660 Da; a default calibration was applied with 13 callibration points. Mass spectra (MS1) were recorded in the reflector positive mode using a laser (wavelength 355 nm) operated at a 200 Hz repetition rate and an accelerated voltage at 2 KV. In a data-dependant acquisition (DDA) mode, MS/MS spectra were acquired for 20 strongest precursors selected between 850 and 4000 Da. A filter of signal-to-noise ratio greater than 20 from one MS scan was for precursor selection. Precursor ions were selected by timed ion selector (TIS), and fragmentation was carried out by collision-induced dissociation (CID) using air as collision gas at 1KV energy and a recharge pressure threshold of 1.5e-006. At least 1200 and 1600 laser shots were accumulated to obtain MS and MS/MS spectra, respectively, and peak list was generated using the 4000 Series Explorer Software v. 3.5 (Applied Biosystem, USA). The peak list was searched against non-redundant protein sequence database of NCBI (NCBInr 25122011, 16338050 sequences). Search parameters were as follows: trypsin digestion with one missed cleavage, variable modifications (oxidation of methionine, and carbamidomethylation of cysteine) and peptide mass tolerance for precursor ions was ±0.6 Da, and the mass tolerance for fragment ions was ±0.3 Da. For all proteins successfully identified by MS/MS, MASCOT score greater than 60 was accepted as significant (p-value < 0.05).
Bioinformatics Tools
Bioinformatics approaches were applied to proteins identified from the Ft LVS. Subcellular protein localization was predicted with PSLpred [2]. Classical signal peptides were detected with SignalP (ver. 4.1) (http://www.cbs.dtu.dk/services/SignalP), and signal peptides of lipoproteins were predicted with LipoP (ver. 1.0) (http://www.cbs.dtu.dk/services/LipoP). Non-classically secreted proteins were predicted with SecretomeP (ver. 2.0) (http://www.cbs.dtu.dk/services/SecretomeP).
Results
Identification of Immunogenic Proteins from F. tularensis LVS
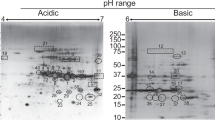
Whole-cell lysate of Ft LVS was separated on 2DE gel (Fig. 1a) and the corresponding Western blot was probed with serum from rabbit immunized with LVS (NCTC 10857). Representative Western blot is shown in Fig. 1b with a total of 38 immunogenic protein spots in the pH range 5–8, corresponding to 28 unique proteins identified by MALDI-TOF/TOF analysis (Table 1). Multiple charge variants for some of the proteins were observed, and for most of the proteins, the identification was based on the MS–MS analysis of two or more matched peptides with significant scores in the Mascot database search (Table S1). Specific details of MS/MS analysis are given in supplementary Table S1. As the protein identification was based on an ion search at the NCBI non-redundant database in the taxonomic group of Bacteria (16,338,050 entries), chances of false-positive hits are substantially reduced. The MW and pI values of the protein spots on the 2-DE gels were compared with the theoretical MW and pI values of corresponding proteins from Francisella (Table S1). It showed a close match on most of the occasions that further indicates unambiguous identification of proteins. A few differences between theoretical and experimental values could be attributed to post-translational proteolytic processing and modification, cleavage of alkaline regions, and the phosphorylation of multiple residues. Serum from immunized rabbit showed intense reactivity with the outer membrane protein FopA. Other proteins of LVS strain that showed immunoreactivity included peptidoglycan-associated lipoprotein, intracellular growth locus, acetyl-CoA carboxylase, malonyl CoA-acyl carrier protein transacylase, ABC transporter, ATP-binding protein, rhodanese, elongation factor Tu, elongation factor EF-Ts, elongation factor G, dihydrolipoamide acetyltransferase, fumarate hydratase, UDP-glucose/GDP-mannose dehydrogenase, ClpB protein, chitinase family 18 protein, glutamate dehydrogenase, chaperonin protein DnaK, chaperonin protein groEL, inosine-5′-monophosphate dehydrogenase, malate dehydrogenase, peroxidase/catalase, aspartyl/glutamyl-tRNA amidotransferase, cell division protein, dihydrolipoamide dehydrogenase, acetyl-CoA acetyltransferase, and succinyl-CoA synthetase. In addition, three hypothetical proteins FTL_1225, FTL_0572, and FTL_0637 also indicated immunoreactivity with Anti-Ft LVS serum.

Coomassie-stained 2D reference gel (a) and representative 2D immunoblot (b) showing immunoreactive proteins of F. tularensis LVS in pH range 5–8. Table 1 refers identified immunogenic protein spots indicated by numbers
Bioinformatic Analysis of Immunoreactive Proteins
Properties of the immunogenic proteins were examined according to computationally predicted features to determine whether a particular type of protein was overrepresented. The identified proteins were classified according to the Clusters of Orthologous Groups (http://www.ncbi.nlm.nih.gov/COG/) where the identified proteins were arranged according to predicted function. Pie chart shows that majority of the proteins belonged to the category of energy production and conversion (14%); translation, ribosomal structure, and biogenesis (14%); and post-translational modification, protein turnover, and chaperones (14%) (Fig. 2a). This was followed by 11% of the identified proteins predictably involved in the cell wall, membrane, and envelop biogenesis. PSLpred method was used to predict the subcellular localization of proteins based on various features of proteins such as amino acid sequence, dipeptide composition, physiochemical properties, and information of PSI-Blast. Graphical Fig. 2b shows that the majority of identified immunoreactive proteins were predicted to be cytoplasmic in location (71%), followed by extracellular (11%), outer membrane (7%), inner membrane (4%), and periplasmic (7%) proteins.

Graphical representation of a clusters of orthologous groups (COGs) and b predicted subcellular location using PSLpred method, for identified immunoreactive proteins (color figure online)
Among these immunoreactive antigens, two (peptidoglycan-associated lipoprotein and hypothetical protein FTL_1225) were predicted as lipoproteins and three carried signal peptidase I-cleavage sites (hypothetical protein FTL_0572, chitinase, and outer membrane protein FopA). Nine proteins (peptidoglycan-associated lipoprotein, dihydrolipoamide acetyltransferase, HP FTL_1225, rhodanese, molecular chaperone DnaK, chitinase, peroxidase/catalase, outer membrane protein FopA, and HP FTL_0572) are predictably secreted through nonclassical secretory mechanism.
Discussion
Tularemia is a zoonosis that has gained scientific attention after 2001 anthrax attack, and concerns about the possible future use of type A F. tularensis as a weapon of mass destruction have renewed the interest in developing antitularemia vaccines [28]. Currently, no vaccine has been licensed for use to prevent the cases of tularemia, and use of F. tularensis LVS as a vaccine alternative is plagued with several limitations. Adaptive immune responses, either antibody-centric or T cell dominated, may complement, augment, and regulate each other in case of Francisella infection [6]. The subunit vaccine for F. tularensis is not available due to the lack of identified Ft antigens capable of inducing protection against the highly virulent type A Ft strains. The selection of suitable animal model represents a weak point in anti tularemia vaccine development. Mice continue to be exploited for vaccination studies, but they are more sensitive to primary pulmonary infection with F. tularensis than humans. Therefore, their use for evaluation of primary mechanisms of Francisella pathogenesis and immune response for vaccine studies is not appropriated. It is necessary to combine several animal models to confirm the potential benefit of an experimental vaccine for humans, especially the one which closely mimics pathogenesis and immune response mechanisms [24].
Tularemia is also identified as rabbit fever because rabbits are vectors for the disease, but the spectrum of immunogenic molecules of F. tularensis in activation of rabbit immune system is poorly understood. In this study, the protein spots found reactive to rabbit Anti-Ft LVS serum, by immunoblotting-linked gel image analysis of Ft LVS proteins, were selected. Using the tandem mass spectrometry, 38 immunogenic protein spots corresponding to 28 proteins showing immunoreactivity were identified. The immunodominant proteins of Ft LVS comprise proteins expressed from genes encoding for diverse functions including energy production and conversion; and translation, post-translational modification, and chaperones. The outer membrane protein FopA showed intense reactivity with serum from Ft LVS-immunized rabbit. FopA is observed to be immunogenic in many of the immunoproteomic studies pertaining to pathogenic bacteria [3, 12, 14, 15, 17, 30, 32, 33]. Chaperone proteins DnaK and GroEL (60 kDa) have not only been described as T cell reactive antigens, but also as molecules inducing a specific antibody response in human Francisella infection [17]. The chaperones GroEL and DnaK are found immunogenic in many pathogens including Streptococcus pyogenes, Burkholderia pseudomallei, and Brucella abortus [12]. DnaK, GroEL, FopA, and peroxidase/ catalase (KatG) identified here failed to provide protective immunity against virulent type A F. tularensis infection in a previous report using recombinant antigens [1].
The immuno-recognition of nine proteins identified here (ClpB protein, cell division protein FtsZ, elongation factor Tu, outer membrane protein FopA, peroxidase/catalase, chaperonin GroEL, molecular chaperone DnaK, acetyl-CoA carboxylase, biotin carboxyl carrier protein, dihydrolipoamide dehydrogenase, and intracellular growth locus), has been demonstrated in the tularemia infection of rabbit, mice, and humans [3, 12, 14–18, 23, 29, 30, 32, 33]. Twelve antigens in the current study have been previously reported to be immunogenic in the human humoral response to LVS (Table 1). GroEL has also been found immunodominant antigen in non-human primate, while acetyl-CoA carboxylase, biotin carboxyl carrier protein subunit (accB), has immunoreactivity in broad range of hosts including non-human primate and rat other than mice and human [4]. Glutamate dehydrogenase and chaperonin protein DnaK have been identified as immunoreactive with sera from the Type A tularemia patients [14]. Five immunogenic proteins (chaperone protein groEL, Acetyl-CoA carboxylase, biotin carboxyl carrier protein, chaperone protein DnaK, outer membrane-associated protein FopA, and elongation factor EF-Ts) identified in this study were also detected previously by protein microarray approach using sera against F. tularensis [12].
Thirteen proteins from this study are predictably surface-localized, of which seven (FopA, Acetyl-CoA carboxylase, biotin carboxyl carrier protein, elongation factor Tu, DnaK, peroxidase/catalase, chitinase, and GroEL) were found to be immunoreactive and primary candidates for a defined post-exposure vaccine [3]. Eight new Ft LVS immunogenic proteins observed in the present study using rabbit model include fumarate hydratase, dihydrolipoamide acetyltransferase, acetyl-CoA acetyltransferase, hypothetical proteins (FTL_0637, FTL_0572, FTL_1225), aspartyl/glutamyl-tRNA amidotransferase, and rhodanese (Table 1). Fumarate hydratases participate in a wide range of cellular processes including TCA cycle, helps in energy production, and was reported as putative target for the development of antileishmanial drugs [13]. Fumarate hydratase was also observed to increase in expression under in vivo growth conditions of F. tularensis [30]. Dihydrolipoamide acetyltransferase along with dihydrolipoamide dehydrogenase is component of pyruvate dehydrogenase (PDH) complex which was found to be immunogenic in Bacillus anthracis and Bacillus thuringiensis [8]. The PDH complex used in the TCA cycle is highly immunogenic in other bacterial species, such as Neisseria meningitidis, Mycoplasma capricolum, and Mycoplasma hyopneumoniae. Recently, PDH has been tested as a DNA vaccine against Mycoplasma mycoides subsp. mycoides, the causative agent of contagious bovine pleuropneumonia [21]. Acetyl-CoA acetyltransferase has been found to be immunogenic in Burkholderia multivorans and Burkholderia cenocepacia, during Human Infection [27] and in Clostridium perfringens ATCC 13124 in case of murine infection [26]. Notably, rhodanese protects Pseudomonas aeruginosa from cyanide toxicity and increases in expression after in vitro exposure of F. tularensis strain FSC033 to hydrogen peroxide [5, 30]. Lipoproteins have been successfully used as the potential protective antigens against some pathogens [26]; thus, peptidoglycan-associated lipoprotein and hypothetical protein FTL_1225 present attractive targets for the evaluation of their vaccine potential against tularemia. As other animal models of tularemia are developed and characterized, there exists opportunities to correlate the profile of immunoreactive proteins generated by LVS vaccination with the protective status of the host animal. The immunogenic Francisella proteins identified in this study might provide supporting information for developing strong vaccine candidates that can elicit an effective and specific immune response. Cell-mediated immune response to these antigens is required to be characterized to further validate their efficacy as subunit vaccine candidates.
To the best of knowledge, this study is the first report of humoral immune response in rabbit against heat-killed Ft LVS. We report here eight new Ft LVS immunogenic proteins using rabbit model [fumarate hydratase, dihydrolipoamide acetyltransferase, acetyl-CoA acetyltransferase, hypothetical proteins (FTL_0637, FTL_0572, FTL_1225), aspartyl/glutamyl-tRNA amidotransferase, and rhodanese]. Keeping in mind the subtle advantages of subunit vaccine over live attenuated vaccines, these proteins offer novel putative vaccine candidates for tularemia infection. A detailed immunological characterization for these candidates will further be helpful in screening the candidates for challenge studies. Together with the previously described immunoreactive antigens in mouse and human model, the novel immunogenic proteins in rabbit model described here will be helpful to validate new target antigens for the generation of a subunit or DNA vaccine for this medically and militarily important pathogen.
References
Banik S, Mansour AA, Suresh RV, WykoffClary S, Malik M, McCormick AA, Bakshi CS (2015) Development of a multivalent subunit vaccine against tularemia using Tobacco Mosaic Virus (TMV) based delivery system. PLoS ONE 10(6):e0130858. doi:10.1371/journal.pone.0130858
Bhasin M, Garg A, Raghava GP (2005) PSLpred: prediction of subcellular localization of bacterial proteins. Bioinformatics 21(10):2522–2524
Chandler JC, Sutherland MD, Harton MR, Molins CR, Anderson RV, Heaslip DG, Bosio CM, Belisle JT (2015) Francisella tularensis LVS surface and membrane proteins as targets of effective post-exposure immunization for tularemia. J Proteome Res 14(2):664–675. doi:10.1021/pr500628k
Chu P, Cunningham AL, Yu J-J, Nguyen JQ, Barker JR, Lyons CR, Wilder J, Valderas M, Sherwood RL, Arulanandam BP, Klose KE (2014) Live attenuated Francisella novicida vaccine protects against Francisella tularensis pulmonary challenge in rats and non-human primates. PLoS Pathog 10(10):e1004439. doi:10.1371/journal.ppat.1004439
Cipollone R, Frangipani E, Tiburzi F, Imperi F, Ascenzi P, Visca P (2007) Involvement of Pseudomonas aeruginosa rhodanese in protection from cyanide toxicity. Appl Environ Microbiol 73(2):390–398. doi:10.1128/AEM.02143-06
Cowley SC, Elkins KL (2011) Immunity to Francisella. Front Microbiol 2:26. doi:10.3389/fmicb.2011.00026
De Pascalis R, Mittereder L, Chou AY, Kennett NJ, Elkins KL (2015) Francisella tularensis vaccines elicit concurrent protective T- and B-Cell immune responses in BALB/cByJ mice. PLoS ONE 10(5):e0126570. doi:10.1371/journal.pone.0126570
Delvecchio VG, Connolly JP, Alefantis TG, Walz A, Quan MA, Patra G, Ashton JM, Whittington JT, Chafin RD, Liang X, Grewal P, Khan AS, Mujer CV (2006) Proteomic profiling and identification of immunodominant spore antigens of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Appl Environ Microbiol 72(9):6355–6363. doi:10.1128/AEM.00455-06
Dennehy R, McClean S (2012) Immunoproteomics: the key to discovery of new vaccine antigens against bacterial respiratory infections. Curr Protein Pept Sci 13(8):807–815
Dennis DT, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Friedlander AM, Hauer J, Layton M, Lillibridge SR, McDade JE, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Tonat K (2001) Tularemia as a biological weapon: medical and public health management. JAMA 285:2763–2773. doi:10.1001/jama.285.21.2763
Ellis J, Oyston PC, Green M, Titball RW (2002) Tularemia. Clin Microbiol Rev 15:631–646. doi:10.1128/CMR.15.4.631-646.2002
Eyles JE, Unal B, Hartley MG, Newstead SL, Flick-Smith H, Prior JL, Oyston PC, Randall A, Mu Y, Hirst S, Molina DM, Davies DH, Milne T, Griffin KF, Baldi P, Titball RW, Felgner PL (2007) Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics 7(13):2172–2183. doi:10.1002/pmic.200600985
Feliciano PR, Gupta S, Dyszy F, Dias-Baruffi M, Costa-Filho AJ, Michels PAM, Nonato MC (2012) Fumarate hydratase isoforms of Leishmania major: subcellular localization, structural and kinetic properties. Int J Biol Macromol 51(1–2):25–31. doi:10.1016/j.ijbiomac.2012.04.025
Fulton KM, Zhao X, Petit MD, Kilmury SLN, Wolfraim LA, House RV, Sjostedt A, Twine SM (2011) Immunoproteomic analysis of the human antibody response to natural tularemia infection with Type A or Type B strains or LVS vaccination. Int J Med Microbiol 301(7):591–601. doi:10.1016/j.ijmm.2011.07.002
Havlasova J, Hernychova L, Brychta M, Hubalek M, Lenco J, Larsson P, Lundqvist M, Forsman M, Krocova Z, Stulik J, Macela A (2005) Proteomic analysis of anti-Francisella tularensis LVS antibody response in murine model of tularemia. Proteomics 5(8):2090–2103. doi:10.1002/pmic.200401123
Havlasova J, Hernychova L, Halada P, Pellantova V, Krejsek J, Stulik J, Macela A, Jungblut PR, Larsson P, Forsman M (2002) Mapping of immunoreactive antigens of Francisella tularensis live vaccine strain. Proteomics 2(7):857–867
Janovska S, Pavkova I, Hubalek M, Lenco J, Macela A, Stulik J (2007) Identification of immunoreactive antigens in membrane proteins enriched fraction from Francisella tularensis LVS. Immunol Lett 108(2):151–159. doi:10.1016/j.imlet.2006.12.004
Janovska S, Pavkova I, Reichelova M, Hubaleka M, Stulik J, Macela A (2007) Proteomic analysis of antibody response in a case of laboratory acquired infection with Francisella tularensis subsp. tularensis. Folia Microbiol 52(2):194–198
Kirimanjeswara GS, Olmos S, Bakshi CS, Metzger DW (2008) Humoral and cell-mediated immunity to the intracellular pathogen Francisella tularensis. Immunol Rev 225:244–255. doi:10.1111/j.1600-065X.2008.00689.x
Kumar B, Alam SI, Kumar O (2013) Host response to intravenous injection of epsilon toxin in mouse model: A proteomic view. Proteomics 13:89–107. doi:10.1002/pmic.201200227
March JB, Jepson CD, Clark JR, Totsika M, Calcutt MJ (2006) Phage library screening for the rapid identification and in vivo testing of candidate genes for a DNA vaccine against Mycoplasma mycoides subsp. mycoides small colony biotype. Infect Immun 74:167–174. doi:10.1128/IAI.74.1.167-174.2006
Oyston PC (2009) Francisella tularensis vaccines. Vaccine 27(Suppl 4):D48–D51. doi:10.1016/j.vaccine.2009.07.090
Pelletier N, Raoult D, La Scola B (2009) Specific recognition of the major capsid protein of Acanthamoeba polyphaga mimivirus by sera of patients infected by Francisella tularensis. FEMS Microbiol Lett 297:117–123. doi:10.1111/j.1574-6968.2009.01675.x
Putzova D, Senitkova I, Stulik J (2016) Tularemia vaccines. Folia Microbiol. doi:10.1007/s12223-016-0461-z
Reed DS, Smith L, Dunsmore T, Trichel A, Ortiz LA, Cole KS, Barry E (2011) Pneumonic tularemia in rabbits resembles the human disease as illustrated by radiographic and hematological changes after infection. PLoS ONE 6(9):e24654. doi:10.1371/journal.pone.0024654
Sengupta N, Alam SI, Kumar B, Kumar RB, Gautam V, Kumar S, Singh L (2010) Comparative proteomic analysis of extracellular proteins of Clostridium perfringens type A and type C strains. Infect Immun 78(9):3957–3968. doi:10.1128/IAI.00374-10
Shinoy M, Dennehy R, Coleman L, Carberry S, Schaffer K, Callaghan M, Doyle S, McClean S (2013) Immunoproteomic analysis of proteins expressed by two related pathogens, Burkholderia multivorans and Burkholderia cenocepacia, during human infection. PLoS ONE 8(11):e80796. doi:10.1371/journal.pone.0080796
Sunagar R, Kumar S, Franz BJ, Gosselin EJ (2016) Tularemia vaccine development: paralysis or progress? Vaccine. 6: p. 9–23. doi:10.2147/VDT.S85545
Sundaresh S, Randall A, Unal B, Petersen JM, Belisle JT, Hartley MG, Duffield M, Titball RW, Davies DH, Felgner PL, Baldi P (2007) From protein microarrays to diagnostic antigen discovery: a study of the pathogen Francisella tularensis. Bioinformatics 23(13):i508–i518. doi:10.1093/bioinformatics/btm207
Twine SM, Mykytczuk NC, Petit M, Shen H, Sjostedt A, Conlan JW, Kelly JF (2006) In vivo proteomic analysis of the intracellular bacterial pathogen, Francisella tularensis, isolated from mouse spleen. Biochem Biophys Res Commun 345(4):1621–1633. doi:10.1016/j.bbrc.2006.05.070
Twine SM, Mykytczuk NC, Petit M, Tremblay TL, Conlan JW, Kelly JF (2005) Francisella tularensis proteome: low levels of ASB-14 facilitate the visualization of membrane proteins in total protein extracts. J Proteome Res 4(5):1848–1854
Twine SM, Petit MD, Fulton KM, House RV, Conlan JW (2010) Immunoproteomics analysis of the murine antibody response to vaccination with an improved Francisella tularensis live vaccine strain (LVS). PLoS ONE 5(4):e10000. doi:10.1371/journal.pone.0010000
Twine SM, Petit MD, Shen H, Mykytczuk NC, Kelly JF, Conlan JW (2006) Immunoproteomic analysis of the murine antibody response to successful and failed immunization with live anti-Francisella vaccines. Biochem Biophys Res Commun 346(3):999–1008. doi:10.1016/j.bbrc.2006.06.008
Acknowledgements
Authors thank Director DRDE Gwalior for providing all facilities and support required for this study. Authors are also thankful to High Containment Facility and Animal Facility of DRDE for providing animal. The first author acknowledges the Grant of fellowship by Defence Research and Development Organization, Government of India.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
All Authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gaur, R., Alam, S.I. & Kamboj, D.V. Immunoproteomic Analysis of Antibody Response of Rabbit Host Against Heat-Killed Francisella tularensis Live Vaccine Strain. Curr Microbiol 74, 499–507 (2017). https://doi.org/10.1007/s00284-017-1217-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-017-1217-y