
Abstract
Although gut microbiota has been studied relatively extensively in the context of allergic diseases, there have been several contradictions between these studies. By applying high-throughput sequencing, we aimed to analyze the differences in gut microbiota between atopic and healthy children at 5 and 12 years of age. 51 stool samples were collected from 14 atopic and 15 healthy children and analyzed with 454 pyrosequencing of the 16S rRNA gene. At the ages of 5 and 12 years, Bacteroides, Prevotella, and Dialister dominated gut microbiota in both atopic and healthy groups of children. Children in the atopic group had lower abundance and prevalence of Akkermansia in gut microbiota than their healthy counterparts. Thus, the composition of gut microbiota does not seem to be significantly different between atopic and healthy children, but lower abundance and prevalence of Akkermansia indicate that this bacterium may accompany or play a role in IgE-mediated atopic diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Normal gut microbiota is essential for digestive processes such as fermentation of carbohydrates, but it also takes part in immunological processes by participating in the development of gut-associated lymphoid tissues (GALTs) and providing resistance to pathogens [12, 26]. Dysbiosis of gut microbiota has been linked to several metabolic and immunological disorders such as diabetes, irritable bowel syndrome, and allergies [13, 16, 20].
Correlation between the composition of gut microbiota and manifestation of an allergic disease has been studied relatively extensively throughout the years, and most of the studies have observed significant differences between the gut microbiota of healthy and allergic individuals, but the exact nature of these differences has yet to be defined [20]. Several studies have linked decreased levels of bifidobacteria [14, 22], increased levels of clostridia [6, 11, 22], and Enterobacteriaceae [18] to allergy, but others have failed to confirm these reports [7, 14, 20]. The limitations of applied methodologies may be one of the reasons behind contradictory results as these have rarely been high throughput and simultaneously quantitative and qualitative. Only a few studies analyzing the composition of gut microbiota in the context of allergy have used high-throughput sequencing [1, 2], which enables to both identify different bacteria and estimate their relative abundance [25]. The broader application of this methodology may give a better understanding of the differences in gut microbiota existing between allergic and healthy individuals.
By applying high-throughput 454 pyrosequencing of the 16S rRNA gene, we aimed to comprehensively analyze the differences in composition of gut microbiota between 12-year-old atopic and healthy children and also determine the differences in the composition at the age of 5 years when many of the children were not yet sensitized to allergens.
Materials and Methods
Participants and Sampling
14 atopic (10 male) and 15 healthy (7 male) 12-year-old participants were selected from a larger study of immune response development to allergens and the development of allergy in relation to environmental factors [24]. Stool samples (approximately 1–2 g) were collected from the participants at 5 and 12 years of age. Samples collected at both time points (at 5 and 12 years of age) were available for seven children from the atopic and 15 from the healthy group of participants. Altogether, 51 stool samples were available, 22 from children at 5 years of age (7 belonging to atopic and 15 to healthy group) and 29 from children at 12 years of age (14 and 15, respectively).
The children were divided into atopic and healthy group based on their sensitization at 12 years of age. The atopic group comprised children who were sensitized to at least 1 allergen detected with a positive skin prick test (SPT) and had detectable circulating allergen-specific IgE antibodies at 12 years of age. At 5 years of age, majority of the children in atopic group were not yet sensitized to tested allergens. The healthy group comprised children with no sensitization to any tested allergens nor any clinical manifestation of an allergic disease at any age. Atopic sensitization was detected using SPT and IgE measurements. SPT tests were performed in duplicate on the volar aspects of the forearms, using lancets of ALK (HØrsholm, Denmark) with standardized extracts of inhalant allergens (cat, dog, house dust mite, birch, timothy, mugwort from Solu-Prick SQTM, ALK; cockroach from Bayer AG), histamine hydrochloride (10 mg/ml) as a positive, and glycerol as a negative control. The SPT was considered positive when the mean of the longest and right angle diameters of one of the wheals was at least 3 mm. For IgE measurements, a commercial chemiluminescence method UniCap was used to measure IgE antibodies of inhalant allergens (cat, dog, house dust mite, cockroach, birch, timothy grass, mugwort) from plasma according to the recommendations of the manufacturer Pharmacia & Upjohn Diagnostics AB.
The research Ethics Committee of the University of Tartu approved the study. Parents or guardians and also participants themselves signed an informed consent agreement.
DNA Extraction and 454 Sequencing of the 16S rRNA Gene
DNA was extracted from stool samples using a QIAamp DNA stool mini kit (Qiagen N.V.) with some modifications. 1 g of feces was suspended in 10 ml of PBS buffer and homogenized. 0.3 g of 0.1 mm zirconia/silica beads and 1.4 ml of ASL solution from the stool mini kit were added to 1 ml of pellet cells. The tubes were then agitated for 3 min at a speed of 5000 rpm in a mini-bead beater (Biospec Products Inc.). The protocol was then continued as described by the manufacturer.
The amplification of 16S rRNA V1–V2 hypervariable region was carried out with two sequential PCR reactions with second PCR extending the length of the amplicon derived from the first PCR. Reaction volume was 25 μL, including pfu DNA polymerase (Thermo Scientific Inc.), 3 μL of DNA template (for second PCR, 50 times diluted amplicon deriving from the first PCR was used), and primers at concentration 0.2 μM. Primer sequences and cycling parameters are described in Table 1. PCR products were purified using Agencourt AMPure XP (Beckman Coulter Inc.) and sequenced with 454 FLX+ systems at GATC Biotech AG.
Data and Statistical Analysis
MOTHUR software 1.27.0 was used to trim, denoise, and align the sequences; generate OTUs; and assign taxonomy. OTUs were generated with the average neighbor hierarchical clustering algorithm with an identity threshold of 97 %. For additional de-noising, OTUs with less than two sequences were removed. Taxonomic assignments were performed using Ńaive Bayesian classifier with a confidence cutoff of 100 % against aligned 16S rRNA gene reference sequences obtained from the SILVA ribosomal RNA database. The relative abundance values and sequence counts were all normalized.
Statistical analysis and graphics were done using R 2.13.2 software. Categorical values were compared with the Fisher exact, and the continuous variables with Wilcoxon rank sum test. Three linear regression analyses were used to analyze the relationship between genera Bacteroides and Prevotella in relation to the most abundant genera (mean relative abundance >0.005), the Shannon diversity index, sampling time, and atopic sensitization. All regression analyses were carried out with the Holm-Bonferroni correction. The level of a significant difference was set at 5 %.
Results and Discussion
To our knowledge, this is the first study applying high-throughput sequencing to analyze the composition of gut microbiota in 5- and 12-year-old atopic and healthy children. Our results indicate that the manifestation of IgE-mediated atopy in childhood is not accompanied by major alterations in the adult-like gut microbiota at 5 and 12 years of age as the composition of gut microbiota in both groups was dominated by the same taxa with similar abundance. The only exception was genus Akkermansia, which is significantly less prevalent and abundant in the gut microbiota of atopic children. These results are in conflict with several earlier studies and indicate that the differences in the gut microbiota accompanying atopy may not be as extensive as previously thought.
We received altogether 194 841 high-quality sequence reads (with an average length of 261 bp). The cutoff for the minimal number of trimmed sequences assigned to individual samples was set to 1600 (at that level, rarefaction curves for 98 % of the samples reached a 5 % plateau; Supplementary Fig. 1). 49 of the initial 51 samples and 1614 OTUs crossed this cutoff value and were used for further analysis (GenBank accession numbers for representative sequences of operational taxonomic units (OTUs) KM975951–KM977564). Retrieved OTUs were distributed between 7 phyla, 34 families, and 56 genera and were mainly identified at the family or genus level.
The gut microbiota in atopic and healthy children was not significantly different based on Cosine similarity index values [mean (SD) Cosine similarity index between samples collected from atopic versus healthy children was 0.4 (0.33); within the atopic group the similarity was 0.39 (0.34) and in the healthy group 0.43 (0.33)]. Samples collected from the same individuals at 5 and 12 years of age were more similar to each other than to other samples [mean (SD) Cosine similarity index for samples collected from the same individuals was 0.59 (0.32) versus the mean (SD) similarity with other samples 0.4 (0.33); P value = 0.02]. Also, bacterial diversity did not differ significantly between atopic and healthy children being on average (SD) 2.3 (0.6). Based on OTU composition, there was no clustering of samples observed in the principal component analysis (PCA) plot (Fig. 1).

Two-dimensional plot of the first two PCA components describing the variance of OTU composition between samples collected from atopic and healthy 5- and 12-year-old children
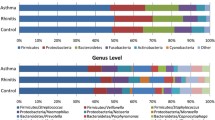
Lack of significant differences between the abundance values of taxa dominating the gut microbiota in atopic and healthy children can be observed in Table 2, which describes mean (SD) relative abundance values of dominating taxa (relative abundance ≥0.005) in atopic and healthy children at 5 and 12 years of age. The taxonomic composition in both atopic and healthy children was similar to the findings of earlier studies analyzing normal gut colonization process in adults being dominated by Bacteroidetes (specifically genus Bacteroides) and Firmicutes [3].
Interestingly, linear regression analysis showed negative correlation between the abundance of Bacteroides and the abundance of Prevotella, Dialister (P values < 0.001), and Shannon diversity index (P value <0.001; Adjusted R 2 = 0.82). Also, the abundance of Prevotella was negatively correlated with the abundance of Bacteroides and Dialister (P values <0.001 and P = 0.004, respectively) and Shannon diversity index (P = 0.01; Adjusted R 2 = 0.74). Negative correlation between the abundance of Bacteroides and Prevotella is in agreement with several other studies analyzing gut microbial composition using metagenomic approaches [3, 15]. These differences have also been described specifically among children as recent studies have described the dominance of Bacteroides rather than Prevotella within the phylum Bacteroidetes in Western children and the opposite in children from Bangladesh and Burkina Faso [8, 17].
Regardless of these differences, there was no correlation observed between the abundance of Bacteroides or Prevotella with the atopic sensitization.
We did not observe increased levels of clostridia, Enterobacteriaceae or staphylococci in atopic children, although these taxa have been proposed to be potential candidates in increasing the risk of atopic sensitization [20]. Clostridia were detected in all samples and the mean (SD) relative abundance was 0.15 (0.07) in atopic and 0.16 (0.1) in healthy children. Enterobacteria were detected in 70 % of the samples with mean (SD) relative abundance 0.01 (0.03) in atopic and 0.004 (0.007) in healthy children. We did not detect any Staphylococcus OTUs. Also, in contrast to a previous study by Sepp et al. [22], we did not observe lower levels of bifidobacteria in atopic children when compared to their healthy counterparts. Bifidobacteria were present in over 90 % of the samples in both groups, and the differences in the abundance of bifidobacteria between these groups were not statistically significant (Table 2). This may be due to the low sensitivity of culturing techniques used by Sepp et al. as they reported bifidobacteria only in 21 % of the samples [22]. We also failed to observe differences in the OTU composition between atopic and healthy children as we detected altogether 13 bifidobacterial OTUs and eight of them were observed in both groups, these included the most abundant bifidobacterial OTUs, which did not differ between groups. This somewhat contradicts the results of Štšepetova et al. [23], who reported the presence of different bifidobacterial species composition in allergic and healthy children, but as OTUs are taxonomical units that in our study were identified mostly at genus level, we cannot confirm that they represent species. Thus, we are unable to confront the results of Štšepetova et al. with full certainty.
Our observations support the results of a recent study by Abrahamsson et al. who used high-throughput sequencing to analyze the diversity and composition of gut microbiota in infants who developed allergic diseases by the age of 7 years [2]. They did not observe any taxonomical differences between these infants and infants who did not develop allergic diseases by the age of 7 years. As we observed the lack of difference in 5- to 12-year-old children it would have been interesting to compare our results with their findings in 7-year-olds, but unfortunately, they did not analyze the community composition of gut microbiota and only performed a clinical follow-up at that age. They confirmed some of the earlier reports by observing the correlation between reduced diversity of gut microbiota during infancy and several allergies manifesting later in childhood [2, 5], which indicates the possibility that the differences in bacterial diversity of gut microbiota during infancy may precede IgE-mediated atopy experienced later in life.
In our study, only the abundance of Akkermansia was significantly lower in atopic than in healthy children both at 5 and 12 years of age (Table 2). The prevalence of genus Akkermansia was also lower in the atopic than in the healthy group at 5 and 12 years of age (29 vs. 93 and 17 vs. 67 %, respectively; P = 0.004 and P = 0.02; Fig. 2). Akkermansia spp., with its mucin-degrading ability, is important for gut barrier function as it regulates the production and thickness of the gut mucus layer [4, 10], but the colonization of Akkermansia in germ-free mice will also lead to transcriptional changes in genes participating in the immune response [9]. It modulates the integrin signaling and antigen presentation pathway in cecum and antigen presentation pathway, B cell receptor signaling, leukocyte extravasation signaling, T cell receptor signaling, IL-4 signaling, and complement and coagulation cascades in the colon [9]. It is not surprising that decreased levels of Akkermansia have been correlated with several inflammatory gut diseases such as irritable bowel syndrome [21], but the effect on the immune system may have a broader scope than only the gastrointestinal tract, as Candela et al. showed lower levels of Akkermansia in 4-14-year-old atopic children than in their healthy counterparts [7]. Our findings are in accordance with Candela et al. and thus it is likely that Akkermansia may accompany or even play a role in IgE-mediated atopic diseases. Whether a lower level of Akkermansia is a causative, resultant or accompanying factor for atopy and what is the exact mechanism behind this correlation remains to be elucidated.

Distribution and abundance of Akkermansia OTUs among participating children. Every column represents the distribution of one Akkermansia OTU (n = 5)
In conclusion, by applying the high-throughput sequencing method, we observed that the gut microbiota of atopic 5- to 12-year-old children is not significantly different from their healthy counterparts, which has recently been reported also in infants. However, we observed lower abundance and prevalence of genus Akkermansia in the gut microbiota of atopic children indicating that Akkermansia may accompany or play a role in IgE-mediated atopic diseases, but the exact mechanism of action needs to be studied further.
References
Abrahamsson TR, Jakobsson HE, Andresson AF, Björksten B, Engstrand L, Jenmalm MC (2011) Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol 129:434–440
Abrahamsson TR, Jakobsson HE, Andersson AF, Björksten B, Engstrand L, Jenmalm MC (2014) Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy. doi:10.1111/cea.12253
Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR et al (2011) Enterotypes of the human gut microbiome. Nature 473:174–180
Belzer C, de Vos WM (2012) Microbes inside- from diversity to function: the case of Akkermansia. ISME J 6:1449–1458
Bisgaard H, Li N, Bonnelykke K, Chawes BLK, Skov T, Paludan-Müller G et al (2011) Reduced diversity of the intestinal microbiota during infancy is associated with increased risk of allergic disease at school age. J Allergy Clin Immunol 128:646–652
Björksten B, Sepp E, Julge K, Voor T, Mikelsaar M (2001) Allergy development and the intestinal microflora during the first year of life. J Allergy Clin Immunol 108:516–520
Candela M, Pampelli S, Turroni S, Severgnini M, Consolandi C, de Bellis G et al (2012) Unbalance of intestinal microbiota in atopic children. BMC Microbiol 12:95
De Filippo C, Cavalieria D, Di Paolab M, Ramazzottic M, Poulletd JB, Massartd S et al (2010) Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. PNAS 107:14691–14696
Derrien M, Baarlen PV, Hooiveld G, Norin E, Müller M, de Vos WM (2011) Modulation of mucosal immune response, tolerance and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front Microbiol 2:166
Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB et al (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. PNAS 110:9066–9071
Kalliomäki M, Kirjavainen P, Eerola E, Kero P, Salminen S, Isolauri E (2001) Distinct patterns of neonatal gut microflora in infants whom atopy was and was not developing. J Allergy Clin Immunol 107:129–134
Kamada N, Seo S-U, Chen GY, Nunez G (2013) Role of the gut microbiota in immunity and inflammatory disease. Nat Rev Immunol 13:321–335
Kassinen A, Krogius-Kurikka L, Mäkivuokko H, Rinttilä T, Paulin L, Corander J et al (2007) The fecal microbiota of irritable bowel syndrome patients differs significantly from that of healthy subjects. Gastroenterology 133:24–33
Kirjavainen PV, Arvola T, Salminen SJ, Isolauri E (2002) Aberrant composition of gut microbiota of allergic infants: a target of bifidobacterial therapy at weaning? Gut 51:51–55
Koren O, Knights D, Gonzalez A, Waldron L, Segata N, Knight R, Huttenhower C, Ley RE (2013) A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput Biol 9:e1002863
Larsen N, Vogensen FK, van den Berg FWJ, Nielsen DS, Andreasen AS, Pedersen BK et al (2010) Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 5:e9085
Lin A, Bik EM, Costello EK, Dethlefsen L, Haque R, Relman DA et al (2013) Distinct distal gut microbiome diversity and composition in healthy children from Bangladesh and the United States. PLoS ONE 8:e53838
Matsumoto M, Ohishi H, Kakizoe K, Benno Y (2004) Faecal microbiota and secretory immunoglobulin A levels in adult patients with atopic dermatitis. Microbiol Ecol 16:13–17
McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z et al (2008) The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog 4:e20
Penders J, Stobberingh EE, van de Brandt PA, Thijs C (2007) The role of the intestinal microbiota in the development of atopic disorders. Allergy 62:1223–1236
Png CW, Linden SK, Gilshenan KS, Zoentendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin THJ (2010) Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 105:2420–2428
Sepp E, Julge K, Mikelsaar M, Björksten B (2005) Intestinal microbiota and immunoglobulin E responses in 5-year-old Estonian children. Clin Exp Allergy 35:1141–1146
Štšepetova J, Sepp E, Julge K, Vaughan E, Mikelsaar M, de Vos WM (2007) Molecularly assessed shifts of Bifidobacterium ssp. and less diverse microbial communities are characteristic of 5-year-old allergic children. FEMS Immunol Med Microbiol 51:260–269
Voor T, Julge K, Böttcher MF, Jenmalm C, Duchén K, Björkstén B (2005) Atopic sensitization and atopic dermatitis in Estonian and Swedish infants. Clin Exp Allergy 35:153–159
Woo PCY, Lau SKP, Teng JLL, Tse H, Yuen K-Y (2008) Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect 14:908–934
Zoentendal EG, Raes J, van de Bogert B, Arumugam M, Booijink GCGM, Troost FJ et al (2012) The human small intestinal microbiota is driven by rapid uptake and conversion of simple carbohydrates. ISME J 6:1415–1426
Acknowledgments
We thank Tiina Rebane from Tartu University Children’s Clinic and Tiiu Rööp from the Institute of Microbiology, University of Tartu for their technical assistance.
The study was supported with a grant from the Estonian Science Foundation (project no. 9180) and Estonian Ministry of Education and Research (target financing No SF0180132s08). Jaak Simm is funded by the KU Leuven Research Council (GOA/10/09 MaNet, CoE PFV/10/016 SymBioSys) and PhD/Postdoc grants from the Flemish Government (IOF, Hercules Stitching, iMinds Medical Information Technologies SBO 2014), by Intel, Janssen Pharmaceutica and by the Institute for the Promotion of Innovation through Science and Technology in Flanders (IWT).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Drell, T., Larionova, A., Voor, T. et al. Differences in Gut Microbiota Between Atopic and Healthy Children. Curr Microbiol 71, 177–183 (2015). https://doi.org/10.1007/s00284-015-0815-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-015-0815-9