
Abstract
Neuromyelitis optica (NMO) is an inflammatory disease that resembles MS in the relapsing clinical course of optic neuritis and myelitis. Two decades of studies have revealed that autoantibodies, reactive to the water channel protein aquaporin 4 (AQP4) are detected in the core group of patients. These autoantibodies play a crucial role in the inflammatory pathology of NMO, involving proinflammatory cytokines, chemokines, and various inflammatory cells such as Th17 cells. Anti-AQP4 antibody–positive NMO differs fundamentally from MS, particularly in the responsiveness to therapies and the neuropathology accompanying destruction of astrocytes. Research into the immunological mechanism has led to the identification of possible targets of therapy, including complement pathway and interleukin-6 (IL-6) receptor signaling. Recent randomized controlled clinical trials have shown the remarkable efficacy of antibodies specific for complement C5, IL-6 receptor, and CD19+ B cells in prevention of NMO spectrum disorder relapses, although no such effects were found in anti-AQP4 antibody–negative patients. These results imply that anti-AQP4 antibody is a biomarker predicting the efficacy of therapies, and indicate the future direction towards “precision medicine.”
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neuromyelitis optica (NMO) is an inflammatory disease of the central nervous system (CNS), characterized by recurrent episodes of optic neuritis and transverse myelitis. Myelitis in NMO tends to have an unusually long, longitudinal lesion in the spinal cord, which may extend to ≥ 3 vertebral segments on magnetic resonance imaging (MRI). The MRI-based description, referred to as longitudinal extensive transverse myelitis, is characteristic for NMO. Unlike multiple sclerosis (MS), NMO rarely has a chronic progressive course. However, disease flare, either at onset or during relapse, is often more serious in NMO than in MS. In fact, the occurrence of even a single relapse may cause persistent disabling symptoms in NMO, implying that prevention of relapse has a significant meaning in NMO. In 2004, Lennon et al. discovered NMO-specific immunoglobulin (Ig) G. The NMO-Ig was reactive to an astrocyte antigen and detected in most NMO patients [1]. Subsequently, the NMO-Ig was found to recognize the water channel protein aquaporin 4 (AQP4), highly expressed in astrocytes [2]. Experts soon realized how remarkable are differences between MS and AQP4-antibody (Ab)-positive NMO. Notably, AQP4-Ab is not only a surrogate marker, but has a pathogenic activity, causing destruction of astrocytes.
Although diagnosis of NMO was based on the clinical observations before an assay for AQP4-Ab was available, the diagnostic criteria established in 2006 [3] considered both the presence of AQP4-Ab and a long-spinal cord lesion. The criteria were very useful for diagnosis of core group of AQP4-Ab-positive NMO. However, the revised criteria put forward in 2015 recommended a broader diagnostic term, NMO spectrum disorder (NMOSD), which includes AQP4-Ab-negative patients as well [4]. As a reliable AQP4-Ab assay is available only in some regions of the world, clinical neurologists have generally supported this recommendation. However, a proportion of anti-AQP4-Ab-negative NMOSD patients have autoantibodies against myelin-oligodendrocyte glycoprotein (MOG). As patients with anti-MOG antibodies (MOG-Ab-positive autoimmune disease) were found to show clinically broader disease endotypes, including cortical and subcortical lesions [5], the outer boundary of NMOSD has become blurred. Moreover, it is now known that AQP4-Ab-positive and AQP4-Ab-negative patients differentially respond to therapies. As such, further research and discussion are needed on the identity of seronegative NMOSD, although AQP4-Ab-positive NMO, on which we focus in this review, is a solid disease entity.
Serious acute relapses in patients with NMOSD are treated with intravenous high-dose corticosteroids and plasma exchanges, as has been performed in MS. However, disease-modifying therapies (DMTs) approved for MS are not recommended for NMOSD, as the use of some DMTs may exacerbate NMOSD [6,7,8]. Thus, oral corticosteroids and generic immunosuppressants are often prescribed in the remission phase of NMOSD. However, recent research has identified various therapeutic targets in the pathogenesis of NMOSD, leading to the development of therapeutic monoclonal antibodies. The efficacy of these antibodies has been demonstrated in randomized clinical trials (RCTs) [9,10,11,12,13], which has opened a gate for new era of NMO therapy.
Clinical features of NMO
The prevalence of NMO is estimated to be 2–4 per 100,000 individuals worldwide [14,15,16]. The median age of disease onset is 39 years, and approximately 90% of the patients are women [16]. It has been reported that familial occurrence of NMO is approximately 3% but the underlying genetic susceptibility is complex [17]. The association of human leukocyte antigen (HLA) with an increased risk of NMO has been revealed in several studies of small size, which reported, for example, the NMO risk of Asian people carrying HLA-DPB1*0501 has been reported [18]. However, the small sample sizes owing to the low prevalence of NMOSD, have limited genome-wide association studies with sufficient power.
NMO is often associated with other autoimmune diseases [19], such as systemic lupus erythematosus and Sjögren’s syndrome, for which genetic factors have been identified. Thus, NMO is presumed to be a multifactorial genetic disease, involving many genes in the immune system. With regard to studies on an environmental factor, it is reported that 15–25% of patients with NMO experience infections prior to disease onset [20, 21]. Several case reports have suggested an association between varicella-zoster virus, human cytomegalovirus, or Epstein-Barr virus [22] and NMO; however, there seems to be no definite pathogen linked with NMO.
To diagnose seropositive NMOSD patients, in addition to AQP4-Ab, at least one of the six core characteristics of this disease, including optic neuritis, acute myelitis, area postrema syndrome (unexplained hiccups, nausea, or vomiting), acute brainstem syndrome, symptomatic narcolepsy or acute diencephalic syndrome, and symptomatic cerebral syndrome, needs to be confirmed. The exclusion of alternative diagnoses is also required [23]. The brainstem manifestations include vomiting, hiccups, oculomotor dysfunction, and pruritus as major symptoms. Hearing loss, facial palsy, vertigo or vestibular ataxia, trigeminal neuralgia, and other cranial nerve signs have also been reported [24]. Symptomatic narcolepsy is characterized by the involvement of the hypothalamus, which expresses high levels of AQP4 [25]. Other manifestations of circumventricular organ involvement include anorexia (hypothalamus), inappropriate antidiuresis, and posterior pituitary endocrinopathy [26]. Notably, these regions are also consistent with AQP4 expression sites in the CNS, especially in the area postrema lying at the base of the fourth ventricle, which has convoluted capillaries that lack tight endothelial junctions. Thus, it seems that anti-AQP4 antibodies directly react to AQP4 on astrocytes forming the glia limitans of the blood–brain barrier, and thereby causing the destruction of these astrocytes [27]. Hemispheric cerebral white matter lesions and peri-ependymal lesions in the diencephalon and white matter adjacent to the lateral ventricles have also been reported as typical brain lesion patterns of NMOSD on MRI, which are interpreted as the core characteristics mentioned above [28]. When AQP4-Ab is negative or a test for AQP4-Ab is not available, diagnosis of seronegative NMOSD is allowed. To make diagnosis of seronegative patients, however, stricter criteria are applied, which include two of the six core clinical characteristics: MRI-confirmed lesions and exclusion of alternative diagnoses [3]. Among the several techniques for detecting AQP4-Ab, cell-based assays have shown the highest sensitivity and specificity compared to other assays, such as enzyme-linked immunosorbent assay. Nevertheless, clinicians should keep in mind that false-negative cases can be diagnosed by repeated AQP4-Ab measures using high-sensitivity techniques [29].
Pathology of NMOSD-complement-dependent astrocytopathy
The advent of AQP4-Ab assays has promoted research into a better understanding of the pathophysiology of NMOSD. Several previous reports have suggested that AQP4-Ab and serum AQP4-Ab titers were correlated with the severity of optic neuritis and with length of the spinal cord lesions [30]. Individual serum AQP4-Ab titers were also found to change in correlation with NMO pathology [31]. However, the titer at onset did not correlate with the number of relapses in the following decade [32]. More fundamentally, histopathological analysis demonstrated that loss of AQP4 expression in astrocytes is associated with complement deposition in the CNS of patients with NMO, which occurs before the destruction of astrocytes [33]. The loss of AQP4 expression appears to be independent of primary demyelination in pathological analysis, further supporting the hypothesis that AQP4-Ab and complement deposition play a pivotal role in the pathogenesis of NMO. Indeed, it has also been reported that levels of glial fibrillary acidic protein (GFAP) in the cerebrospinal fluid (CSF) are remarkably elevated during the relapse of NMO, compared to that in MS, and the activity and severity of NMO may correlate with the levels of GFAP in the blood and CSF, indicating that astrocyte destruction is central to the pathology of NMOSD [34, 35]. Moreover, persistent inflammation leads to continuous recruitment of reactive astrocytes to the lesions wherein astrocytes were destroyed by AQP4-Ab, which causes further neuronal damage in the lesions. Usually, in the tissue repair process of the CNS, reactive astrocytes enter the lesions to form a glial scar; however, in NMO, only a few scattered reactive astrocytes are present in the lesions [36]. Bright spotty lesions on T2-weighted spinal MRI images have been reported as a specific finding of NMO [37], which may reflect this pathological characteristic. The autopsy of an NMO case with long-term immunosuppressive treatment indicated the presence of persistent active inflammatory lesions, suggesting that chronic inflammation may be sustained in at least some patients with NMO [38]. Finally, several reports have shown the pathogenicity of AQP4-Ab in vivo, by showing the enhancement of signs of a murine experimental model experimental autoimmune encephalomyelitis (EAE) after transfer of AQP4-Ab [39,40,41]. The direct pathogenicity of AQP4-Ab has also been proven in a later study by intracerebral injection of both AQP4-Ab and human complement in mice, which recapitulated the histological characteristics of human NMOSD [42]. The result of a pivotal clinical trial showing the efficacy of eculizumab confirmed the role of complement-dependent pathogenesis of AQP4-Ab-positive NMOSD [9].
AQP4 as the autoantigen of NMOSD
AQP4 on perivascular astrocyte end-feet exists as two isoforms, differing at their N termini, due to either translation initiation at the first methionine (M1, 323 aa) or second methionine (M23, 301 aa) [43]. These two isoforms are expressed in the form of heterotetramers that aggregate into the plasma membrane to form two-dimensional supramolecular structures named orthogonal array of particles (OAPs) [44, 45]. M23 and M1 isoforms have opposing effects on the intramembrane organization of AQP4: M23 forms large square arrays with abundant cross-bridges while M1 restricts square array assembly [46]. AQP4 is present not only in brain astrocytes, but also in the eye, ear, skeletal muscle, stomach parietal cells, and kidney collecting ducts [47]. However, tissues expressing AQP4, except for the CNS, are only slightly affected by NMOSD. One possibility is the reduced expression of complement regulator proteins (CD46, CD55, and CD59) [48] in astrocyte foot processes compared with peripheral AQP4-expressing cells, which may be a reason why AQP4-expressing cells in the peripheral organs have increased resistance to complement-dependent cytotoxicity (CDC). In rodents, it has been reported that one of the complement regulators prevents peripheral organ damage, myocarditis, which is caused by intraperitoneal injection of AQP4-Ab in CD59-deficient mice [49]. Another reason could be differences in antigenicity related to the structural difference of AQP4 on astrocytes. AQP4 is expressed in two isoforms, M23 and M1, in a ratio of approximately 3:1 in astrocytes, consisting OAPs assembled from 4- to 6-nm intramembrane particles corresponding to individual AQP4 tetramers. Super-resolution microscopy revealed that recombinant AQP4-Ab with enhanced CDC preferentially formed organized clusters on supramolecular AQP4 OAPs, thereby linking epitope-dependent multimeric assembly with enhanced complement classical pathway, which results in complement component C1q binding and activation [50]. In NMOSD, T and B cells sensitized to supramolecular AQP4 are activated, leading to the production of AQP4-Ab.
Initially, Lennon et al. found this autoantibody reactive to astrocytes, using serum samples from patients with suspected paraneoplastic autoimmunity. Indeed, a number of studies have reported such an immune response to AQP4 as a manifestation of paraneoplastic syndrome mimicking NMO, in patients with tumor tissues expressing AQP4 [51, 52]. In patients without tumors, cross-reactivity to a protein of an intestinal bacterium has been reported [53]. In this study, T cells from patients with NMO demonstrated greater proliferation to AQP4 peptides that have significant homologies to a sequence within Clostridium perfringens adenosine triphosphate–binding cassette transporter permease. These AQP4 peptide–specific T cells were HLA-DR restricted and exhibited CD4+ T helper 17 (Th17) cell polarization. The study thus suggests molecular mimicry is a possible mechanism for triggering NMO.
Immuno-pathogenesis of NMOSD
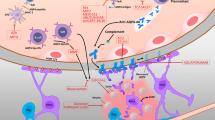
T helper cell subsets are generally assumed to play an important role in inducing adaptive immune responses and serve as initiators of autoimmune diseases. In EAE, an animal model of MS, Th17 cells are defined as a distinct cell lineage and known as pathogenic T cells that produce IL-17 via induction of a proinflammatory cytokine IL-6 receptor (IL-6R) signal followed by activation via IL-23 receptor signaling [54, 55]. Levels of IL-17 in the blood and CSF are elevated in both NMOSD and MS as compared with other non-inflammatory neurological diseases [56]. However, unlike IL-17 that is increased in both NMOSD and MS, elevations of IL-6 and IL-23 are characteristic of NMOSD [56], suggesting that more pathogenic Th17 cells may be induced and operative in the pathology of NMOSD. Of note, circulating follicular-like T helper (cTfh) 17 cells, expressing CXC-chemokine receptor 5 (CXCR5), are reported to strongly support antibody secretion by facilitating differentiation of B cells [57]. Of interest, skewing the cTfh subsets towards cTfh17 was observed in NMOSD [58]. It is important to note that the pathological process of NMO begins with the activation of pathogenic T cells, although this fact is often overlooked or untold as compared with the latter process including anti-AQP4-Ab production, astrocyte injury, and neuronal damage (Fig. 1).

Pathophysiology of NMOSD and key molecule-targeted therapies. This cartoon depicted the dynamics of pathophysiology of NMOSD from autoreactive T cell activation to neuronal damage. T cells in peripheral lymphoid organs are activated by antigen-presenting cells including B cells. IL-6 from B cells may play a pivotal role in differentiating T cells into pathogenic Th17 cells. Along with this, autoreactive B cells differentiat into AQP4-Ab-producing plasmablasts. AQP4-Ab appeared in perivascular space recognize AQP4 on astrocyte end-feet and destruct it as complement-dependent manner. Disruption of blood–brain barrier induces the activation of microglia and dysfunction of astrocytes that yield to the result of neuronal damage. Three key molecule-targeted monoclonal antibodies showed strong effectiveness for preventing relapse of NMOSD: Eculizumab blocks complement C5 cleavage preventing formation membrane attack complex, satralizumab blocks IL-6R signal in multiple cell types, and inebilizumab depletes CD19+ B cells including plasmablasts
IL-6 in NMOSD
In the context of the inflammatory pathology of NMOSD, prior studies have indicated that IL-6 promotes the activation of autoreactive T cells, prolongs the survival of AQP4-Ab-producing B cells, and reduces blood–brain barrier integrity. The results of two pivotal clinical trials showing the efficacy of satralizumab in AQP4-Ab-positive NMOSD have firmly established the role of IL-6 and IL-6R signaling in the pathogenesis of NMO [10, 11].
Pioneering works by Kishimoto et al. initially identified IL-6 as a cytokine that induces B cell production of immunoglobulins [59]. Subsequent studies have identified various roles for IL-6 and demonstrated how this cytokine transmits signals leading to a variety of biological outcomes. IL-6 is a pleiotropic cytokine that induces the synthesis of acute inflammatory phase proteins such as C-reactive protein, serum amyloid A, and fibrinogen from hepatocytes, whereas it inhibits the production of albumin [60]. It also stimulates antibody production and induces the differentiation of effector T cells. Moreover, it promotes the activation and development of some non-immune cells, such as fibroblasts and platelets. Thus, persistent production of IL-6 may trigger the onset or exacerbation of immune-mediated diseases. The binding of IL-6 to cell surface IL-6R forms the IL-6/IL-6R complex, which induces the homodimerization of glycoprotein 130 (gp130). IL-6 binding to soluble IL-6R also induces gp130 homodimerization. This event critically triggers the activation of the Janus kinase (JAK)-STAT3 pathway and JAK-SHP-2-mitogen-activated protein kinase pathway, leading to biological responses.
Previous studies have documented the elevation of various cytokines and chemokines in the serum and CSF of patients with NMOSD [56, 61]. Notably, the levels of the pro-inflammatory IL-6 in the serum of NMOSD were higher than those detected in other neurological diseases, including MS [62, 63]. In the pathogenesis of NMOSD, IL-6 is thought to play multiple roles, including induction of pathogenic effector T cells, promotion of plasmablasts’ survival and their production of AQP4-Ab, and disruption of blood–brain barrier integrity and function [64]. It has been reported that AQP4 peptides from the membrane surface of AQP4 can activate Th17 cells to produce IL-17 [53]. This process could be influenced by IL-6, as this cytokine is critical for the development of Th17 cells, whereas it suppresses regulatory T cell differentiation [65]. Furthermore, IL-6 drives production of IL-21 from human CD4+ T cells, the cytokine capable of inducing Th17 and B cell differentiation [66, 67]. In a rodent model, it has been reported that IL-6 produced by B cells plays a predominant role in inducing Th17 cells associated with brain inflammation [68].
B cells in NMOSD
B cells play multiple roles in the pathophysiology of NMOSD. As B cells produce IL-6, B cell–targeting therapy may show efficacy by reducing the source of the IL-6 involved in NMOSD. Moreover, B cells serve as autoreactive AQP4-Ab producing cells. Consistently, B-cell targeting therapies, such as anti-CD20 and anti-CD19 antibodies, have proven to be efficacious in NMOSD [12, 13].
B cells generated in the bone marrow are immature, precursor B cells that express IgM. These cells further mature into naive B cells and then into follicular B cells or extrafollicular B cells. When activated, the follicular and extrafollicular B cells can differentiate into plasmablasts and short-lived plasma cells, both of which can secrete antibodies. Alternatively, with the help of T helper cells, follicular B cells can also differentiate into memory B cells, which are long-lived, and express antibodies of switched class and high affinity for antigens. When reactivated by antigens, memory B cells can differentiate into plasmablasts, which are competent to become long-lived plasma cells [69].
During an acute NMOSD relapse, dynamic exchange of B cells occurs between the periphery and the CNS. We and others reported that differentiated plasmablasts (CD19lowCD27highCD38highCD180−B cells) are increased in the peripheral blood of patients with NMOSD compared to that in healthy individuals or patients with MS (70, 71). Although the plasmablasts have eccentric nuclei, perinuclear hof regions, and abundant cytoplasm, they still have a larger nucleus with a lower extent of chromatin clumping compared to plasma cells. Thus, it seems that plasmablasts are involved in the process of cell differentiation just before mature plasma cells localized to the bone marrow. Of note, IL-6 was found to promote the survival of plasmablasts derived from patients with active NMOSD. The plasmablasts produced AQP4-Ab ex vivo, and IL-6 enhanced autoantibody production from plasmablasts. These phenomena were not observed when they were stimulated by other B cell–stimulating factors, such as B cell–activating factor and proliferation-inducing ligand [70]. Moreover, blocking IL-6R signaling by an antagonistic antibody for IL-6R decreased the number of cultured plasmablasts.
A small proportion of plasmablasts remains in the secondary lymphoid organ (the spleen or lymph node) where they are generated. Most plasmablasts migrate either to inflamed tissue or bone marrow. In the former case, under the control of interferon-γ-induced expression of CXCR3, they migrate towards CXC-chemokine ligand 9 (CXCL9), CXCL10, and CXCL11. It has been reported that the levels of CXCL10 are increased in the CSF of NMOSD compared to other neurological diseases (69). In the latter case, under the control of chemotaxis towards CXCL12 (which binds CXCR4), plasmablasts are homed towards the bone marrow. All three tissues, inflamed tissue, secondary lymphoid organs, and bone marrow, have finite numbers of plasma cell survival niches. Plasmablasts that succeed in the acquisition of such a niche differentiate into plasma cells and become immobile. Resolution of inflamed tissue after a successful immune response terminates the survival niches in the tissue and therefore eliminates the resident plasma cells, which is the peak of the immune response. In the bone marrow, and to a lesser degree in secondary lymphoid organs, long-lived plasma cells have been shown to survive and provide humoral memory response (66). Importantly, the increase in plasmablasts was more prominent in the relapse phase of NMOSD, especially in the CSF. These plasmablasts in the CSF expressed plasma cell surface protein CD138 and HLA class II. Reflecting their ability to migrate to the CSF, they also expressed inflammatory tissue-oriented CXCR3, but not bone marrow–derived CXCR4 (70). Interestingly, the number of somatic hypermutations did not differ between plasmablasts from peripheral blood and CSF, and some clones were commonly observed between the two sites. Thus, peripherally differentiated plasmablasts probably migrate into the CNS and could be part of inflammation on site during the flare of NMOSD. AQP4-Ab produced from plasmablasts in the CSF has been shown to induce pathogenicity in a rodent model [41].
It is still unclear how autoreactive B cells recognizing AQP4 are able to differentiate into plasmablasts. Self-reactive B cells are usually eliminated either by positive selection in the bone marrow or negative selection in lymphoid follicles. Thus, self-reactive B cells with low affinity for self-antigens can exhibit peripheral circulation. B cells require antigen recognition through B cell receptor for clonal expansion. As most autoantibodies recognize intracellular self-antigens, cross-reactivity to the cell surface or extracellular molecules enhances clonal expansion and differentiation into memory and plasma cells. As a hypothesis, the innate immune response through pattern recognition receptors, such as toll-like receptor (TLR) signals, can activate self-reactive B cells and overcome tolerance [74]. Indeed, immature B cells from patients with AQP4-Ab-positive NMOSD have the potential to produce polyreactive autoantibodies against intracellular antigens compared to healthy controls [75]. The role of extrafollicular B cells and their prominence in severe disease have been implicated in human systemic lupus erythematosus. Its activation is mediated by hyper-responsiveness to TLR-7 and leads to the generation of a unique B cell subpopulation, such as activated naïve B cells, IgD−CD27− double–negative B cells expressing CD11c, and autoreactive antibody-secreting plasmablasts [76]. Double–negative B cells as well as plasmablasts were reported to be increased in patients with NMOSD [71, 77]. TLR-7-driven differentiation of B cells from patients with NMOSD has been shown to facilitate AQP4-Ab production [78]. Therefore, the B cell differentiation pathway commonly found in autoimmune diseases may exist in NMOSD.
Neuroinflammation in NMOSD
Astrocyte foot processes line the perivascular space of the cerebral blood vessels and form part of the blood–brain barrier. As AQP4 is expressed in the foot processes and functions as a water channel, the AQP4-Ab released around blood vessels can easily recognize AQP4 in the astrocytes. In NMOSD pathology, this antigen–antibody reaction causes astrocyte damage at the same site in a complement-dependent manner, resulting in the breakdown of the blood–brain barrier and subsequent CNS inflammation. In an in vitro blood–brain barrier model study, AQP4-mediated astrocyte injury using IgG (NMO-IgG) derived from patients with NMOSD enhanced vascular permeability by reducing the barrier function of vascular endothelial cells. It has been suggested that NMO-IgG may promote the influx of inflammatory cells into the brain by enhancing the expression of chemokines that attract inflammatory cells to the endothelial cells [79]. In this process, IL-6 produced from astrocytes by NMO-IgG may transmit a signal via the IL-6R in the vascular endothelium. In fact, in animal model, EAE has been reported to ameliorate in vascular endothelial cell–specific IL-6R knockout mice [80]. Additionally, AQP4-Ab may affect co-localized excitatory amino acid transporter 2 (EAAT2) in astrocytes, causing glutamate hyperexcitability at neuronal synapses due to EAAT2 downregulation, resulting in neuronal damage [81]. NMO-IgG has also been shown to induce the production of complement component C3 from astrocytes, which may lead to activation of microglia, induction of perivascular microglial migration, and further exacerbation of local inflammation [82]. Further research is needed to elucidate how astrocyte damage caused by AQP4-Ab leads to neuronal damage, and how complement, inflammatory cytokines, and inflammatory cells, which have been key pathological conditions in previous analyses, are involved in neuroinflammation in the CNS.
Molecular targeted therapies in NMOSD
As in the case of MS, intravenous high-dose corticosteroid therapy and plasma exchange therapy are used for the treatment of acute NMOSD. However, DMTs for MS are not recommended for preventing NMOSD relapses due to their potentials to exacerbate NMOSD [6,7,8]. Therefore, oral corticosteroids and common immunosuppressive agents, such as azathioprine, mycophenolate mofetil, methotrexate, tacrolimus, and cyclosporine A [83,84,85,86,87], have been prescribed as preventive agents for the recurrence of NMOSD. As a molecular targeted drug, the efficacy of rituximab targeting the B cell surface receptor CD20 has been described [88], and a randomized controlled trial (RCT) in Japan confirmed its safety and usefulness [13]. Moreover, recent advances in this field have identified three key molecules as therapeutic targets in the molecular pathway of NMOSD, and the effectiveness of monoclonal antibody drugs targeting complement C5, IL-6R, and B cell surface molecule CD19 has been demonstrated in RCTs [9,10,11,12] (Table 1).
Complement protein C5–targeted therapy: Eculizumab
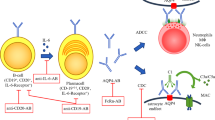
Eculizumab, a humanized monoclonal antibody, inhibits the terminal complement protein C5 and prevents its cleavage into C5a, which is proinflammatory, and C5b, which coordinates the formation of the membrane attack complex (MAC) [89]. In general, antibody-mediated target cell damage includes CDC and antibody-dependent cellular cytotoxicity (ADCC) exerted by natural killer cells and other cells with similar functions. Eculizumab inhibits C5 to C5b cleavage in the former pathway, which is essential for the formation of a MAC at the end of the classical complement pathway that begins with antibody-antigen recognition. Preclinical data indicate that AQP4-Ab triggers the complement cascade both in vitro and in vivo [42, 90], which leads to inflammation and MAC formation. MAC is implicated in astrocyte destruction and subsequent neuronal injury, which is absent in experimental models treated with a complement inhibitor [91]. The role of eculizumab in preventing C5a formation is likely to prevent several mediators of inflammation, such as phagocytosis and vascular permeability [92]. In vitro experiments have shown that C5a-preactivated neutrophils mediate the formation of reactive astrocytes in the sera of patients with NMOSD [93].
In an initial open-label study involving 14 patients with clinically active AQP4-Ab-positive NMOSD, the use of eculizumab reduced the frequency of relapse [94]. Thus, a phase III RCT (PREVENT study) was conducted to examine the recurrence prevention effect of 143 AQP4-Ab-positive NMOSD patients recruited from 18 countries (70 institutes), and a marked reduction in relapse was observed in the treatment group compared to that in the placebo group. The study was terminated early because of its strong inhibitory effect (hazard ratio 0.06; 95% CI, 0.02 to 0.20) [9]. Eculizumab may cause serious infections with capsule-forming bacteria, such as Neisseria meningitidis, of which eradication needs the action of MAC. Thus, vaccination against Neisseria meningitidis is required before starting therapy, and clinicians need to closely observe the conditions of the patients after the start of treatment by bi-weekly injection of the reagent. In a recent study assessed, the long-term efficacy and safety of eculizumab treatment of NMOSD for 3.7 years. Along with a low relapse rate (0.025 relapses/person-year), no patient developed a meningococcal infection, although there were some serious infections [95]. The efficacy of this therapeutic agent provided the most significant evidence that astrocyte damage in CNS lesions occurs in a complement-dependent manner.
IL-6R signal–targeted therapy: Satralizumab
Satralizumab is a humanized monoclonal antibody that binds to the membrane-bound and soluble forms of IL-6R to block the homodimerization of gp130, thereby preventing downstream signaling pathways. Satralizumab is designed to dissociate from IL-6R at low pH in the endosome, and to maintain the affinity of constant regions for the neonatal Fc receptor (FcRn). This engineering allows the antibody to dissociate from IL-6R after internalization of the antigen–antibody complex in the target cells followed by its release into the circulation. This results in binding to another IL-6R (recycling mechanism) that prolongs the elimination half-life of the drug in the blood by a monthly subcutaneous injection during the maintenance period [10, 96]. Increased levels of IL-6 were detected in the serum or CSF of patients with NMOSD [61, 62], and preclinical studies have shown its association with pathogenic T cells, B cells, vascular endothelial cells, and astrocytes [53, 70, 79]. In particular, blocking IL-6R signaling decreased the number of cultured AQP4-Ab-producing plasmablasts, which prompted us to further study the treatment of NMOSD by IL-6R blockade. Case reports from Japan and Germany have indicated that tocilizumab, a humanized monoclonal antibody against IL-6R, could be valuable for treatment-resistant patients [97, 98]. A Japanese patient who was previously treated with interferon-β showed gradual recovery from severe neuropathic pain, fatigue, and disability scores [97]. Subsequently, a small study exploring the safety and efficacy of tocilizumab confirmed the efficacy of this treatment, indicating that IL-6R signaling plays a crucial role in the pathogenesis of NMOSD [99]. In Germany, tocilizumab showed efficacy in patients with intractable NMOSD who were resistant to intensive treatment with steroids, immunosuppressants such as anti-CD20, and plasmapheresis [98]. Recently, a clinical trial with tocilizumab for NMOSD, including 85% seropositivity for AQP4-Ab, was performed as monotherapy. In this study, a higher percentage of patients without concurrent autoimmune diseases remained relapse-free for 2 years than those treated with azathioprine (91% vs. 63%) [100].
The efficacy and safety of subcutaneous satralizumab treatment concomitant with the administration of baseline immunosuppressants were tested in a phase III RCT (SAkuraSky study) [10]. A total of 83 patients with NMOSD (both seropositive and seronegative for AQP4-Ab) were recruited from 11 countries (34 institutes). Of note, both adult patients (18–74 years) and adolescents (12–17 years) were included in the study. As baseline immunosuppressants, concomitant use of azathioprine, mycophenolate mofetil, or prednisolone was allowed. The doses of the baseline treatment drugs were fixed during the study. In the AQP4-Ab seropositive subgroup, the effect of satralizumab was more prominent than that of the patients receiving placebo (hazard ratio, 0.21; 95% CI, 0.06–0.75). In striking contrast, there was only a small difference between the satralizumab and placebo groups in the AQP4-Ab seronegative group. This may reflect the difference in pathological conditions between the AQP4-Ab seropositive and seronegative groups. In this study, based on previous experience in case series, pain or fatigue was assessed as a secondary outcome; however, there was no significant difference between the groups regarding the pain score or the fatigue score from baseline to week 24, which requires further assessment in a post-marketing surveillance. The safety was similar between the adult and adolescent groups. Adverse events in the satralizumab group, including injection-related reactions, nasopharyngitis, upper respiratory tract infections, and headaches, were also observed in the placebo group. The efficacy and safety of subcutaneous satralizumab treatment as monotherapy were also tested in a phase III RCT (SAkuraStar study) [11]. A total of 95 patients with NMOSD (both seropositive and seronegative for AQP4-Ab) were recruited from 13 countries (44 institutes). The randomization strategy included previous therapy for relapse prevention, whether or not the patients had been treated with B cell–depleting therapy or not. In the AQP4-Ab seropositive group, the effect of satralizumab was more prominent than in the patients receiving placebo (hazard ratio, 0.26; 95% CI, 0.11–0.63). In contrast, there was no difference between the satralizumab and placebo groups in the AQP4-Ab−seronegative group. This study revealed that satralizumab monotherapy in patients with NMOSD delayed the occurrence of relapse and reduced the relapse rate in AQP4-Ab seropositive patients. The long-term efficacy and safety of these two trials have been announced by the pharmaceutical company with consistent adverse events in double-blind periods for 4 years. As patients with NMOSD often have other autoimmune diseases, the use of tocilizumab in other autoimmune diseases may provide clinicians with a sense of side effect prediction, such as tuberculosis. The effect of this therapeutic agent reaffirmed the importance of the IL-6R signaling at each checkpoint in the pathology of NMOSD.
CD19 + B cell–targeted therapy: Inebilizumab
Inebilizumab is a humanized, affinity-optimized, afucosylated monoclonal antibody that binds to the B-cell surface antigen CD19. Compared to anti-CD20 monoclonal antibodies that recognize and deplete a small subset of CD20-expressing T lymphocytes (in addition to B lymphocytes) [101], anti-CD19 antibodies recognize and deplete a wider range of lymphocytes exclusively from the B cell lineage, including CD20-negative precursor B cells and plasmablasts [102]. The efficacy and safety of inebilizumab as monotherapy were tested in a phase II/III RCT (N-MOmentum study). A total of 99 patients with NMOSD (both seropositive and seronegative for AQP4-Ab) were recruited from 25 countries. This therapy was administered twice: first at intervals of 2 weeks and then at intervals of 6 months. The number of CD20-positive B cells in the blood continued to be depleted from the 8th day after administration. In the AQP4-Ab seropositive group, the effect of inebilizumab was more prominent than that in the patients receiving placebo (hazard ratio, 0.272; 95% CI, 0.150–0.496). On the other hand, three relapses were observed only in the inebilizumab group of 13 patients with AQP4-Ab seronegative NMOSD compared to the placebo group of four seronegative NMOSD patients. Eculizumab is injected intravenously every 2 weeks and satralizumab is injected subcutaneously every month. As so, the long interval of inebilizumab injection can reduce the burden on patients. However, as with other therapies, clinicians should be careful about infectious diseases. Rituximab has been reported to cause fulminant hepatitis due to viral reactivation in hepatitis B carrier patients [103]. The long-term efficacy and safety of inebilizumab treatment of NMOSD for more than 4 years were assessed in patients with AQP4-Ab seropositive NMOSD. Along with a low relapse rate (0.052 relapses/person-year) especially from 2nd years, IgG levels decreased over time [104]. The suppression of the recurrence of NMOSD by treatment targeting B cells in the circulating blood indicates that the process from lymphocyte activity to tissue damage often progresses dynamically with each recurrence in the pathology of NMOSD.
Conclusion
Since the discovery of AQP4-Ab, studies on the pathophysiology of NMOSD have been performed logically and intensively. It might be possible to state that the sequences of research into NMOSD have been a most successful endeavor to elucidate immune pathogenesis of neurological diseases. In fact, RCTs have shown the efficacy of therapeutic agents targeting complement, IL-6R signaling, and B cells, and these drugs have become available in many countries, including the USA, Canada, and Japan. With regard to the practice, we need to be able to recommend proper use of a drug in individual cases. The new molecular targeted drugs efficiently prevent relapses, which obviously reduces the burden of patients. However, NMOSD patients are suffering from continuous neurological signs, including pain, fatigue, and unstable visual acuity. It was previously thought that these problems could be solved only partially by medicine. However, pathological studies have begun to reveal the presence of chronic inflammation in NMOSD, which may account for the persistent symptoms. The next goal of research may be how much we can reduce the persistent symptoms caused by chronic inflammation.
References
Lennon VA, Wingerchuk DM, Kryzer TJ, Pittock SJ, Lucchinetti CF, Fujihara K, Nakashima I, Weinshenker BG (2004) A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 364:2106–2112
Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR (2005) IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 202:473–477
Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG (2006) Revised diagnostic criteria for neuromyelitis optica. 2006. Neurology 66:1485–1489
Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG, International Panel for NMOD (2015) International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85:177–89
Reindl M, Di Pauli F, Rostasy K, Berger T (2013) The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 9:455–461
Warabi Y, Matsumoto Y, Hayashi H (2007) Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J Neurol Sci 252:57–61
Izaki S, Narukawa S, Kubota A, Mitsui T, Fukaura H, Nomura K (2013) A case of neuromyelitis optica spectrum disorder developing a fulminant course with multiple white-matter lesions following fingolimod treatment. Rinsho Shinkeigaku 53:513–517
Jacob A, Hutchinson M, Elsone L, Kelly S, Ali R, Saukans I, Tubridy N, Boggild M (2012) Does natalizumab therapy worsen neuromyelitis optica? Neurology 79:1065–1066
Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, Nakashima I, Terzi M, Totolyan N, Viswanathan S, Wang KC, Pace A, Fujita KP, Armstrong R, Wingerchuk DM (2019) Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 381:614–625
Yamamura T, Kleiter I, Fujihara K, Palace J, Greenberg B, Zakrzewska-Pniewska B, Patti F, Tsai CP, Saiz A, Yamazaki H, Kawata Y, Wright P, De Seze J (2019) Trial of satralizumab in neuromyelitis optica spectrum disorder. N Engl J Med 381:2114–2124
Traboulsee A, Greenberg BM, Bennett JL, Szczechowski L, Fox E, Shkrobot S, Yamamura T, Terada Y, Kawata Y, Wright P, Gianella-Borradori A, Garren H, Weinshenker BG (2020) Safety and efficacy of satralizumab monotherapy in neuromyelitis optica spectrum disorder: a randomised, double-blind, multicentre, placebo-controlled phase 3 trial. Lancet Neurol 19:402–412
Cree BAC, Bennett JL, Kim HJ, Weinshenker BG, Pittock SJ, Wingerchuk DM, Fujihara K, Paul F, Cutter GR, Marignier R, Green AJ, Aktas O, Hartung HP, Lublin FD, Drappa J, Barron G, Madani S, Ratchford JN, She D, Cimbora D, Katz E, N-MOmentum study inverstigators (2019) Inebilizumab for the treatment of neuromyelitis optica spectrum disorder (N-MOmentum): a double-blind, randomised placebo-controlled phase 2/3 trial. Lancet 394:1352–1363
Tahara M, Oeda T, Okada K, Kiriyama T, Ochi K, Maruyama H, Fukaura H, Nomura K, Shimizu Y, Mori M, Nakashima I, Misu T, Umemura A, Yamamoto K, Sawada H (2020) Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol 19:298–306
Cossburn M, Tackley G, Baker K, Ingram G, Burtonwood M, Malik G, Pickersgill T, te Water Naude J, Robertson N (2012) The prevalence of neuromyelitis optica in South East Wales. Eur J Neurol 19:655–9
Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO (2011) A population-based study of neuromyelitis optica in Caucasians. Neurology 76:1589–1595
Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG (2007) The spectrum of neuromyelitis optica. Lancet Neurol 6:805–815
Matiello M, Kim HJ, Kim W, Brum DG, Barreira AA, Kingsbury DJ, Plant GT, Adoni T, Weinshenker BG (2010) Familial neuromyelitis optica. Neurology 75:310–315
Yoshimura S, Isobe N, Matsushita T, Yonekawa T, Masaki K, Sato S, Kawano Y, Kira J, South Japan Multiple Sclerosis Genetics C (2013) Distinct genetic and infectious profiles in Japanese neuromyelitis optica patients according to anti-aquaporin 4 antibody status. J Neurol Neurosurg Psychiatry 84:29–34
Pittock SJ, Lennon VA, de Seze J, Vermersch P, Homburger HA, Wingerchuk DM, Lucchinetti CF, Zephir H, Moder K, Weinshenker BG (2008) Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 65:78–83
Wingerchuk DM, Hogancamp WF, O’Brien PC, Weinshenker BG (1999) The clinical course of neuromyelitis optica (Devic’s syndrome). Neurology 53:1107–1114
Ghezzi A, Bergamaschi R, Martinelli V, Trojano M, Tola MR, Merelli E, Mancardi L, Gallo P, Filippi M, Zaffaroni M, Comi G, Italian Devic’s Study G (2004) Clinical characteristics, course and prognosis of relapsing Devic’s neuromyelitis Optica. J Neurol 251:47–52
Sellner J, Hemmer B, Muhlau M (2010) The clinical spectrum and immunobiology of parainfectious neuromyelitis optica (Devic) syndromes. J Autoimmun 34:371–379
Amiry-Moghaddam M, Ottersen OP (2003) The molecular basis of water transport in the brain. Nat Rev Neurosci 4:991–1001
Kremer L, Mealy M, Jacob A, Nakashima I, Cabre P, Bigi S, Paul F, Jarius S, Aktas O, Elsone L, Mutch K, Levy M, Takai Y, Collongues N, Banwell B, Fujihara K, de Seze J (2014) Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler 20:843–847
Kanbayashi T, Shimohata T, Nakashima I, Yaguchi H, Yabe I, Nishizawa M, Shimizu T, Nishino S (2009) Symptomatic narcolepsy in patients with neuromyelitis optica and multiple sclerosis: new neurochemical and immunological implications. Arch Neurol 66:1563–1566
Zekeridou A, Lennon VA (2015) Aquaporin-4 autoimmunity. Neurol Neuroimmunol Neuroinflamm 2:e110
Shosha E, Dubey D, Palace J, Nakashima I, Jacob A, Fujihara K, Takahashi T, Whittam D, Leite MI, Misu T, Yoshiki T, Messina S, Elsone L, Majed M, Flanagan E, Gadoth A, Huebert C, Sagen J, Greenberg BM, Levy M, Banerjee A, Weinshenker B, Pittock SJ (2018) Area postrema syndrome: frequency, criteria, and severity in AQP4-IgG-positive NMOSD. Neurology 91:e1642–e1651
Wang KY, Chetta J, Bains P, Balzer A, Lincoln J, Uribe T, Lincoln CM (2018) Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder: a pictorial review. Br J Radiol 91:20170690
Waters PJ, McKeon A, Leite MI, Rajasekharan S, Lennon VA, Villalobos A, Palace J, Mandrekar JN, Vincent A, Bar-Or A, Pittock SJ (2012) Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 78:665–71
Takahashi T, Fujihara K, Nakashima I, Misu T, Miyazawa I, Nakamura M, Watanabe S, Shiga Y, Kanaoka C, Fujimori J, Sato S, Itoyama Y (2007) Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: a study on antibody titre. Brain 130:1235–1243
Jarius S, Aboul-Enein F, Waters P, Kuenz B, Hauser A, Berger T, Lang W, Reindl M, Vincent A, Kristoferitsch W (2008) Antibody to aquaporin-4 in the long-term course of neuromyelitis optica. Brain 131:3072–3080
kaishi T, Takahashi T, Nakashima I, Abe M, Ishii T, Aoki M, Fujihara K (2020) Repeated follow-up of AQP4-IgG titer by cell-based assay in neuromyelitis optica spectrum disorders (NMOSD). J Neurol Sci 410:116671
Misu T, Fujihara K, Kakita A, Konno H, Nakamura M, Watanabe S, Takahashi T, Nakashima I, Takahashi H, Itoyama Y (2007) Loss of aquaporin 4 in lesions of neuromyelitis optica: distinction from multiple sclerosis. Brain 130:1224–1234
Aktas O, Smith MA, Rees WA, Bennett JL, She D, Katz E, Cree BAC, group NMs, the NMsi (2021) Serum glial fibrillary acidic protein: a neuromyelitis optica spectrum disorder biomarker. Ann Neurol 89:895–910
Misu T, Takano R, Fujihara K, Takahashi T, Sato S, Itoyama Y (2009) Marked increase in cerebrospinal fluid glial fibrillar acidic protein in neuromyelitis optica: an astrocytic damage marker. J Neurol Neurosurg Psychiatry 80:575–577
Roemer SF, Parisi JE, Lennon VA, Benarroch EE, Lassmann H, Bruck W, Mandler RN, Weinshenker BG, Pittock SJ, Wingerchuk DM, Lucchinetti CF (2007) Pattern-specific loss of aquaporin-4 immunoreactivity distinguishes neuromyelitis optica from multiple sclerosis. Brain 130:1194–1205
Yonezu T, Ito S, Mori M, Ogawa Y, Makino T, Uzawa A, Kuwabara S (2014) “Bright spotty lesions” on spinal magnetic resonance imaging differentiate neuromyelitis optica from multiple sclerosis. Mult Scler 20:331–337
Fujii C, Itoh K, Saito K, Satoh Y, Makino M, Nakagawa M, Yamaguchi K, Fushiki S, Mizuno T (2018) Persistent microscopic active inflammatory lesions in the central nervous system of a patient with neuromyelitis optica treated with oral prednisolone for more than 40years. eNeurological Sci 11:17-9
Bradl M, Misu T, Takahashi T, Watanabe M, Mader S, Reindl M, Adzemovic M, Bauer J, Berger T, Fujihara K, Itoyama Y, Lassmann H (2009) Neuromyelitis optica: pathogenicity of patient immunoglobulin in vivo. Ann Neurol 66:630–643
Kinoshita M, Nakatsuji Y, Kimura T, Moriya M, Takata K, Okuno T, Kumanogoh A, Kajiyama K, Yoshikawa H, Sakoda S (2009) Neuromyelitis optica: passive transfer to rats by human immunoglobulin. Biochem Biophys Res Commun 386:623–627
Bennett JL, Lam C, Kalluri SR, Saikali P, Bautista K, Dupree C, Glogowska M, Case D, Antel JP, Owens GP, Gilden D, Nessler S, Stadelmann C, Hemmer B (2009) Intrathecal pathogenic anti-aquaporin-4 antibodies in early neuromyelitis optica. Ann Neurol 66:617–629
Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC (2010) Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain 133:349–361
Jung JS, Bhat RV, Preston GM, Guggino WB, Baraban JM, Agre P (1994) Molecular characterization of an aquaporin cDNA from brain: candidate osmoreceptor and regulator of water balance. Proc Natl Acad Sci U S A 91:13052–13056
Jin BJ, Rossi A, Verkman AS (2011) Model of aquaporin-4 supramolecular assembly in orthogonal arrays based on heterotetrameric association of M1–M23 isoforms. Biophys J 100:2936–2945
Nicchia GP, Cogotzi L, Rossi A, Basco D, Brancaccio A, Svelto M, Frigeri A (2008) Expression of multiple AQP4 pools in the plasma membrane and their association with the dystrophin complex. J Neurochem 105:2156–2165
Furman CS, Gorelick-Feldman DA, Davidson KG, Yasumura T, Neely JD, Agre P, Rash JE (2003) Aquaporin-4 square array assembly: opposing actions of M1 and M23 isoforms. Proc Natl Acad Sci U S A 100:13609–13614
Takata K, Matsuzaki T, Tajika Y (2004) Aquaporins: water channel proteins of the cell membrane. Prog Histochem Cytochem 39:1–83
Zipfel PF, Skerka C (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9:729–740
Yao X, Verkman AS (2017) Complement regulator CD59 prevents peripheral organ injury in rats made seropositive for neuromyelitis optica immunoglobulin G. Acta Neuropathol Commun 5:57
Soltys J, Liu Y, Ritchie A, Wemlinger S, Schaller K, Schumann H, Owens GP, Bennett JL (2019) Membrane assembly of aquaporin-4 autoantibodies regulates classical complement activation in neuromyelitis optica. J Clin Invest 129:2000–2013
Sudo A, Chihara N, Takenaka Y, Nakamura T, Ueda T, Sekiguchi K, Toda T (2018) Paraneoplastic NMOSD associated with EG junction adenocarcinoma expressing unprotected AQP4. Neurol Neuroimmunol Neuroinflamm 5:e482
Mitsui S, Tanaka Y, Kimura K, Jimbo N, Chihara N, Maniwa Y. 2021. Paraneoplastic neuromyelitis optica spectrum disorder associated with atypical thymic carcinoid: a case report. Ann Thorac Cardiovasc Surg
Varrin-Doyer M, Spencer CM, Schulze-Topphoff U, Nelson PA, Stroud RM, Cree BA, Zamvil SS (2012) Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann Neurol 72:53–64
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133
Melamed E, Levy M, Waters PJ, Sato DK, Bennett JL, John GR, Hooper DC, Saiz A, Bar-Or A, Kim HJ, Pandit L, Leite MI, Asgari N, Kissani N, Hintzen R, Marignier R, Jarius S, Marcelletti J, Smith TJ, Yeaman MR, Han MH, Aktas O, Apiwattanakul M, Banwell B, Bichuetti D, Broadley S, Cabre P, Chitnis T, De Seze J, Fujihara K, Greenberg B, Hellwig K, Iorio R, Jarius S, Klawiter E, Kleiter I, Lana-Peixoto M, Nakashima OK, Palace J, Paul F, Prayoonwiwat N, Ruprecht K, Stuve O, Tedder T, Tenembaum S, Garrahan JP, Aires B, van Herle K, van Pelt D, Villoslada P, Waubant E, Weinshenker B, Wingerchuk D, Wurfel J, Zamvil S (2015) Update on biomarkers in neuromyelitis optica. Neurol Neuroimmunol Neuroinflamm 2:e134
Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, Foucat E, Dullaers M, Oh S, Sabzghabaei N, Lavecchio EM, Punaro M, Pascual V, Banchereau J, Ueno H (2011) Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 34:108–121
Nicolas P, Ruiz A, Cobo-Calvo A, Fiard G, Giraudon P, Vukusic S, Marignier R (2019) The balance in T follicular helper cell subsets is altered in neuromyelitis optica spectrum disorder patients and restored by rituximab. Front Immunol 10:2686
Hirano T, Yasukawa K, Harada H, Taga T, Watanabe Y, Matsuda T, Kashiwamura S, Nakajima K, Koyama K, Iwamatsu A et al (1986) Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 324:73–76
Tanaka T, Narazaki M, Kishimoto T (2014) IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295
Uzawa A, Mori M, Arai K, Sato Y, Hayakawa S, Masuda S, Taniguchi J, Kuwabara S (2010) Cytokine and chemokine profiles in neuromyelitis optica: significance of interleukin-6. Mult Scler 16:1443–1452
Barros PO, Cassano T, Hygino J, Ferreira TB, Centuriao N, Kasahara TM, Andrade RM, Linhares UC, Andrade AF, Vasconcelos CC, Alvarenga R, Marignier R, Bento CA (2016) Prediction of disease severity in neuromyelitis optica by the levels of interleukin (IL)-6 produced during remission phase. Clin Exp Immunol 183:480–489
Uzawa A, Mori M, Sato Y, Masuda S, Kuwabara S (2012) CSF interleukin-6 level predicts recovery from neuromyelitis optica relapse. J Neurol Neurosurg Psychiatry 83:339–340
Fujihara K, Bennett JL, de Seze J, Haramura M, Kleiter I, Weinshenker BG, Kang D, Mughal T, Yamamura T. 2020. Interleukin-6 in neuromyelitis optica spectrum disorder pathophysiology. Neurol Neuroimmunol Neuroinflamm 7
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441:235–238
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448:484–487
Diehl SA, Schmidlin H, Nagasawa M, Blom B, Spits H (2012) IL-6 triggers IL-21 production by human CD4+ T cells to drive STAT3-dependent plasma cell differentiation in B cells. Immunol Cell Biol 90:802–811
Molnarfi N, Schulze-Topphoff U, Weber MS, Patarroyo JC, Prod’homme T, Varrin-Doyer M, Shetty A, Linington C, Slavin AJ, Hidalgo J, Jenne DE, Wekerle H, Sobel RA, Bernard CC, Shlomchik MJ, Zamvil SS (2013) MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J Exp Med 210:2921–37
Radbruch A, Muehlinghaus G, Luger EO, Inamine A, Smith KG, Dorner T, Hiepe F (2006) Competence and competition: the challenge of becoming a long-lived plasma cell. Nat Rev Immunol 6:741–750
Chihara N, Aranami T, Sato W, Miyazaki Y, Miyake S, Okamoto T, Ogawa M, Toda T, Yamamura T (2011) Interleukin 6 signaling promotes anti-aquaporin 4 autoantibody production from plasmablasts in neuromyelitis optica. Proc Natl Acad Sci U S A 108:3701–3706
Kowarik MC, Astling D, Gasperi C, Wemlinger S, Schumann H, Dzieciatkowska M, Ritchie AM, Hemmer B, Owens GP, Bennett JL (2017) CNS aquaporin-4-specific B cells connect with multiple B-cell compartments in neuromyelitis optica spectrum disorder. Ann Clin Transl Neurol 4:369–380
Matsushita T, Tateishi T, Isobe N, Yonekawa T, Yamasaki R, Matsuse D, Murai H, Kira J (2013) Characteristic cerebrospinal fluid cytokine/chemokine profiles in neuromyelitis optica, relapsing remitting or primary progressive multiple sclerosis. PLoS ONE 8:e61835
Chihara N, Aranami T, Oki S, Matsuoka T, Nakamura M, Kishida H, Yokoyama K, Kuroiwa Y, Hattori N, Okamoto T, Murata M, Toda T, Miyake S, Yamamura T (2013) Plasmablasts as migratory IgG-producing cells in the pathogenesis of neuromyelitis optica. PLoS ONE 8:e83036
Suurmond J, Diamond B (2015) Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125:2194–2202
Cotzomi E, Stathopoulos P, Lee CS, Ritchie AM, Soltys JN, Delmotte FR, Oe T, Sng J, Jiang R, Ma AK, Vander Heiden JA, Kleinstein SH, Levy M, Bennett JL, Meffre E, O’Connor KC (2019) Early B cell tolerance defects in neuromyelitis optica favour anti-AQP4 autoantibody production. Brain 142:1598–1615
Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z, Bugrovsky R, Blalock EL, Scharer CD, Tipton CM, Wei C, Lim SS, Petri M, Niewold TB, Anolik JH, Gibson G, Lee FE, Boss JM, Lund FE, Sanz I (2018) Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49(725–39):e6
Ruschil C, Gabernet G, Lepennetier G, Heumos S, Kaminski M, Hracsko Z, Irmler M, Beckers J, Ziemann U, Nahnsen S, Owens GP, Bennett JL, Hemmer B, Kowarik MC (2020) Specific induction of double negative B cells during protective and pathogenic immune responses. Front Immunol 11:606338
Wilson R, Makuch M, Kienzler AK, Varley J, Taylor J, Woodhall M, Palace J, Leite MI, Waters P, Irani SR (2018) Condition-dependent generation of aquaporin-4 antibodies from circulating B cells in neuromyelitis optica. Brain 141:1063–1074
Takeshita Y, Obermeier B, Cotleur AC, Spampinato SF, Shimizu F, Yamamoto E, Sano Y, Kryzer TJ, Lennon VA, Kanda T, Ransohoff RM (2017) Effects of neuromyelitis optica-IgG at the blood-brain barrier in vitro. Neurol Neuroimmunol Neuroinflamm 4:e311
Petkovic F, Lazzarino GP, Engblom D, Blomqvist A (2020) IL-6R expressed on CNS vascular endothelial cells contributes to the development of experimental autoimmune encephalomyelitis in mice. J Neuroimmunol 342:577211
Hinson SR, Roemer SF, Lucchinetti CF, Fryer JP, Kryzer TJ, Chamberlain JL, Howe CL, Pittock SJ, Lennon VA (2008) Aquaporin-4-binding autoantibodies in patients with neuromyelitis optica impair glutamate transport by down-regulating EAAT2. J Exp Med 205:2473–2481
Chen T, Lennon VA, Liu YU, Bosco DB, Li Y, Yi MH, Zhu J, Wei S, Wu LJ (2020) Astrocyte-microglia interaction drives evolving neuromyelitis optica lesion. J Clin Invest 130:4025–4038
Mandler RN, Ahmed W, Dencoff JE (1998) Devic’s neuromyelitis optica: a prospective study of seven patients treated with prednisone and azathioprine. Neurology 51:1219–1220
Jacob A, Matiello M, Weinshenker BG, Wingerchuk DM, Lucchinetti C, Shuster E, Carter J, Keegan BM, Kantarci OH, Pittock SJ (2009) Treatment of neuromyelitis optica with mycophenolate mofetil: retrospective analysis of 24 patients. Arch Neurol 66:1128–1133
Kitley J, Elsone L, George J, Waters P, Woodhall M, Vincent A, Jacob A, Leite MI, Palace J (2013) Methotrexate is an alternative to azathioprine in neuromyelitis optica spectrum disorders with aquaporin-4 antibodies. J Neurol Neurosurg Psychiatry 84:918–921
Tanaka M, Kinoshita M, Tanaka K (2015) Corticosteroid and tacrolimus treatment in neuromyelitis optica related disorders. Mult Scler 21:669
Kageyama T, Komori M, Miyamoto K, Ozaki A, Suenaga T, Takahashi R, Kusunoki S, Matsumoto S, Kondo T (2013) Combination of cyclosporine A with corticosteroids is effective for the treatment of neuromyelitis optica. J Neurol 260:627–634
Damato V, Evoli A, Iorio R (2016) Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol 73:1342–1348
Thomas TC, Rollins SA, Rother RP, Giannoni MA, Hartman SL, Elliott EA, Nye SH, Matis LA, Squinto SP, Evans MJ (1996) Inhibition of complement activity by humanized anti-C5 antibody and single-chain Fv. Mol Immunol 33:1389–1401
Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, Fallier-Becker P, Noell S, Lennon VA (2012) Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes. Proc Natl Acad Sci U S A 109:1245–1250
Duan T, Smith AJ, Verkman AS (2018) Complement-dependent bystander injury to neurons in AQP4-IgG seropositive neuromyelitis optica. J Neuroinflammation 15:294
Horiuchi T, Tsukamoto H (2016) Complement-targeted therapy: development of C5- and C5a-targeted inhibition. Inflamm Regen 36:11
Piatek P, Domowicz M, Lewkowicz N, Przygodzka P, Matysiak M, Dzitko K, Lewkowicz P (2018) C5a-preactivated neutrophils are critical for autoimmune-induced astrocyte dysregulation in neuromyelitis optica spectrum disorder. Front Immunol 9:1694
Pittock SJ, Lennon VA, McKeon A, Mandrekar J, Weinshenker BG, Lucchinetti CF, O’Toole O, Wingerchuk DM (2013) Eculizumab in AQP4-IgG-positive relapsing neuromyelitis optica spectrum disorders: an open-label pilot study. Lancet Neurol 12:554–562
Wingerchuk DM, Fujihara K, Palace J, Berthele A, Levy M, Kim HJ, Nakashima I, Oreja-Guevara C, Wang KC, Miller L, Shang S, Sabatella G, Yountz M, Pittock SJ, Group PS (2021) Long-term safety and efficacy of eculizumab in aquaporin-4 IgG-positive NMOSD. Ann Neurol 89:1088–98
Igawa T, Ishii S, Tachibana T, Maeda A, Higuchi Y, Shimaoka S, Moriyama C, Watanabe T, Takubo R, Doi Y, Wakabayashi T, Hayasaka A, Kadono S, Miyazaki T, Haraya K, Sekimori Y, Kojima T, Nabuchi Y, Aso Y, Kawabe Y, Hattori K (2010) Antibody recycling by engineered pH-dependent antigen binding improves the duration of antigen neutralization. Nat Biotechnol 28:1203–7
Araki M, Aranami T, Matsuoka T, Nakamura M, Miyake S, Yamamura T (2013) Clinical improvement in a patient with neuromyelitis optica following therapy with the anti-IL-6 receptor monoclonal antibody tocilizumab. Mod Rheumatol 23:827–831
Ayzenberg I, Kleiter I, Schroder A, Hellwig K, Chan A, Yamamura T, Gold R (2013) Interleukin 6 receptor blockade in patients with neuromyelitis optica nonresponsive to anti-CD20 therapy. JAMA Neurol 70:394–397
Araki M, Matsuoka T, Miyamoto K, Kusunoki S, Okamoto T, Murata M, Miyake S, Aranami T, Yamamura T (2014) Efficacy of the anti-IL-6 receptor antibody tocilizumab in neuromyelitis optica: a pilot study. Neurology 82:1302–1306
Zhang C, Zhang M, Qiu W, Ma H, Zhang X, Zhu Z, Yang CS, Jia D, Zhang TX, Yuan M, Feng Y, Yang L, Lu W, Yu C, Bennett JL, Shi FD, Investigators TS (2020) Safety and efficacy of tocilizumab versus azathioprine in highly relapsing neuromyelitis optica spectrum disorder (TANGO): an open-label, multicentre, randomised, phase 2 trial. Lancet Neurol 19:391–401
Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, Baranzini SE, Leppert D, von Budingen HC (2014) Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 193:580–586
Krumbholz M, Derfuss T, Hohlfeld R, Meinl E (2012) B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol 8:613–623
Evens AM, Jovanovic BD, Su YC, Raisch DW, Ganger D, Belknap SM, Dai MS, Chiu BC, Fintel B, Cheng Y, Chuang SS, Lee MY, Chen TY, Lin SF, Kuo CY (2011) Rituximab-associated hepatitis B virus (HBV) reactivation in lymphoproliferative diseases: meta-analysis and examination of FDA safety reports. Ann Oncol 22:1170–1180
Rensel M, Zabeti A, Mealy MA, Cimbora D, She D, Drappa J, Katz E. 2021. Long-term efficacy and safety of inebilizumab in neuromyelitis optica spectrum disorder: analysis of aquaporin-4-immunoglobulin G-seropositive participants taking inebilizumab for 4 years in the N-MOmentum trial. Mult Scler: 13524585211047223
Funding
This work was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (20H03562 for N.C.), Practical Research Project for Rare/Intractable Diseases by AMED (20ek0109436h0001 and 21ek0109436h0002 for N.C.)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethical statement
The authors declare that this review article does not contain conduction of any animal experiments or studies involving human participants. The authors wrote the article based on the published information without violating any ethical issues.
Conflict of interest
N.C. received speakers’ honorarium from Alexion Pharmaceuticals, Inc., Chugai Pharmaceutical Co., Ltd., and Mitsubishi Tanabe Pharma Corporation. T.Y. received speakers’ honorarium from Alexion Pharmaceuticals, Inc., Chugai Pharmaceutical Co., Ltd., Roche, and Mitsubishi Tanabe Pharma Corporation. T.Y. served as scientific advisor for Chugai and Roche.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is a contribution to the special issue on: Neuroimmune Interactions in Health and Disease - Guest Editors: David Hafler & Lauren Sansing
Rights and permissions
About this article

Cite this article
Chihara, N., Yamamura, T. Immuno-pathogenesis of neuromyelitis optica and emerging therapies. Semin Immunopathol 44, 599–610 (2022). https://doi.org/10.1007/s00281-022-00941-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-022-00941-9