
Abstract
Rheumatoid arthritis (RA) is a chronic inflammatory disease leading to joint destruction. Cytokines play a key role in its pathogenesis. They contribute to the induction and maintenance of inflammation and thus provide therapeutic targets. Many cytokines are involved in RA, and this review focuses on a few critical ones: tumor necrosis factor (TNF), interleukin (IL)-6, IL-1, IL-17, and GM-CSF. TNF and IL-6 are both well-established targets in RA treatment, and new biologic agents are reaching the market. IL-1 represents a more complex cytokine as results in humans do not reach those in animal models. IL-17 and GM-CSF are cytokines representing new targets either as early treatment or in non-responders to other biologics. The interaction between cytokines and their signaling pathways are the basis for the development of new strategies with small molecules or bispecific antibodies. Clearly, the targeting of cytokines has been a major progress in RA treatment, but many issues remain open. Although remission can be better achieved, reactivation of the disease too often occurs upon treatment discontinuation. Better understanding and targeting of chronicity remains a goal to achieve in the future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease, characterized by a breach of immune regulation in the joint synovial membrane leading to severe damage and destruction of cartilage and bone. This disease does not only cause multiple systemic symptoms, such as fever, anemia, osteoporosis, or muscle weakness but also increases cardiovascular morbidity and risk of some cancers [77, 218]. At the local inflammatory site, namely the joint, disturbances in cytokine and chemokine pathways lead to an immune infiltrate that contributes to the increased proliferation of joint fibroblasts, named synoviocytes, and to the chronicity of inflammation [73, 74, 164].
The critical role of cytokines in many immune processes involved in RA pathogenesis [164] has led to the use of novel therapeutics that target specific cytokines with both biologic agents and small molecules [208, 50]. Even if these biologic agents are currently used as common RA therapies, they are prescribed mostly when conventional therapy has failed [216, 220]. Some of these novel biologic agents are now targeted in the standard treatment of RA patients. Indeed, tumor necrosis factor (TNF) or interleukin-6 (IL-6) receptor inhibitors are well-established RA treatments. An interleukin-1 receptor antagonist (IL-1Ra) also belongs to the Food and Drug Administration (FDA)-approved biologic agents. Other cytokines involved in RA pathogenesis are now the targets of second-generation biologic agents, including IL-17 and GM-CSF inhibitors [50].
There are many cytokines involved in RA pathogenesis. In this review, we focus our interest on a few that are central and targeted in RA treatment (TNF, IL-6, IL-1, IL-17, and GM-CSF). For each cytokine, we will describe its signaling pathway and key functions, then its involvement in RA pathogenesis. Finally, we will focus on current and new inhibitors.
TNF
Signaling pathway and functions
Signaling pathway
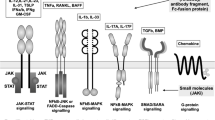
TNF is a pleiotropic cytokine that exerts functions in homeostasis and in pathogenesis of several diseases. TNF is mainly produced by monocytes and macrophages and also by some T cells. The versatile bioactivities of TNF come from the binding to and the activation of two distinct receptors, TNF-receptor 1 (TNFR1) and TNF-receptor 2 (TNFR2) [152, 3]. If TNFR1 is ubiquitously expressed, TNFR2 is restricted to several specific cell types, mainly immune cells, endothelial cells, and others as synoviocytes [70]. The signaling of TNF is activated by a TNF homotrimer that binds a homotrimeric TNFR (Fig. 1). The TNFR1 signaling is activated by soluble and transmembrane TNF while the TNFR2 signaling is activated primarily by transmembrane TNF. TNFR1 carries a death domain that recruits the adaptor protein TNFR1-associated death domain protein (TRADD). This leads to the assembly of different complexes and distinct functional outcomes [24]. TNFR1-complex I activates nuclear factor κB (NFκB) and mitogen-activated protein kinases (MAPKs) that induce inflammation, tissue degeneration, host defense, cell proliferation, and cell survival. TNFR1-complex IIa and b activate caspase-8 that induces apoptosis. TNFR1-complex IIc activates mixed lineage kinase domain-like protein (MLKL) that induces inflammation and necroptosis [132]. TNFR2 does not have a death domain and thus is unable to directly induce programmed cell death. TNFR2 recruits TNFR-associated factor 2 (TRAF2) to form TNFR2-complex I. This complex activates MAPKs, NFκB and protein kinase B (PKB also known as AKT) that induce the same functions as TNFR1-complex I.

Signaling pathways of TNF can bind two receptors, TNFR1 and TNFR2, and its signaling requires a homotrimer. TNFR1 recruits the protein TRADD to its death domain, lacking in TNFR2, and this leads to MLKL and caspase 8 activation mediating cell death. The recruitment of TRAF2 leads to the activation of other pathways (MAPK, NFκB, or PKB), mediating the different functions of TNF. IL-6 signaling goes through two ways. Classic signaling is mediated by mIL-6R and trans-signaling by sIL-6R. Both mechanisms lead to the activation of JAK which promotes STAT or Ras-MAPK activation, inducing the IL-6 functions. IL-1 can bind two receptors. IL-1R1 signaling goes through MyD88 that activates IRAK4. Then, IRAK1 which is associated with TRAF6 is activated by IRAK4, leading to the activation of NFκB and AP-1. IL-1R2 acts as a decoy receptor and thereby IL-1 binding does not induce signal transduction. The binding of IL-1Ra on IL-1R also does not induce signal transduction. IL-17 binds a heterodimeric receptor composed by IL-17RA and IL-17RC. Their conserved signaling domain SEFIR recruits the ubiquitin ligase Act-1. In turn, Act-1 recruits TRAF6 leading to the activation of NFκB, MAPK, and PI3K. GM-CSF binds a heterodimeric receptor, composed by two chains, GM-CSFRα and GM-CSFRβ that activates JAK2. This leads to the activation of STAT5 or PI3K, NFκB, and MAPK pathways, promoting GM-CSF functions
Biological functions of TNF
One model of TNFR signaling suggests that TNFR1 mainly promotes inflammation and tissue degradation while TNFR2 primarily mediates local homeostatic effects such as cell survival or tissue regeneration [132, 202], giving to TNF its versatile bioactivities.
Homeostatic functions
TNF is a key cytokine in inflammation. An optimal defense against pathogens requires TNF. TNF plays a crucial role in host immunity against Mycobacterium tuberculosis (TB) infection. The control of TB infection requires the formation of granulomas, which are highly organized aggregates of macrophages and Th1 cells, and TNF is mandatory to the formation and maintenance of granulomas [172]. In line with this effect in defense, TNF is also involved in organogenesis and development of lymphoid organ architecture and germinal center formation. This cytokine plays a role in the induction of tissue repair, such as neuronal remyelination, cardiac remodeling, or cartilage regeneration [132]. Furthermore, TNF contributes to the resolution of inflammation by inducing apoptosis of inflammatory cells and could also play a role in the inhibition of tumorigenesis through cytostatic effects such as senescence [224, 21]. As mentioned, the homeostatic potential of TNF is at least partially mediated by TNFR2 [132].
Pathogenic functions
One of the major pathogenic functions of TNF is its inflammatory role associated with its chronic uncontrolled production. It induces the production of pro-inflammatory mediators by many cells. For example, TNF induces IL-6 production by synoviocytes from RA patients [145]. Furthermore, TNF orchestrates tissue recruitment and survival of inflammatory immune cells and promotes tissue destruction [72]. TNF plays also a role in autoimmunity, mainly through the inhibition of regulatory T cells [32]. In addition, TNF has a critical role in cardiovascular diseases. Indeed, TNF is cardiotoxic for healthy myocardium by inducing the apoptosis of cardiomyocytes [104]. During tumorigenesis, depending on the inflammatory environment, TNF is able to switch from tumor-inhibition to induction of genotoxic effects and to promotion of tumor-cell survival and proliferation and tumor metastasis [66]. TNF has been involved in the nociceptive response, which is the physiological process underlying the sensation of pain. Indeed, in arthritic patients, the blockade of TNF reduces the increased nociception before inducing anti-inflammatory effects [110].
Thus, TNF is clearly a major pro-inflammatory cytokine, and its uncontrolled production or function has been linked to the pathogenesis of many different inflammatory diseases, such as RA, inflammatory bowel disease, or psoriasis [5].
TNF and RA
Involvement in the pathogenesis
TNF is clearly a central cytokine in RA pathophysiology [73, 74, 164, 23, 132]. TNF displays a wide variety of effector functions relevant to the pathogenesis of RA and thus has a pro-inflammatory influence at several levels. TNF induces leukocyte and endothelial activation, synoviocyte activation and survival, cytokine and chemokine amplification, angiogenesis, and nociceptor activation [164, 23, 163]. In cultures of synovial cells from RA patients, the blockade of TNF significantly decreases the production of other pro-inflammatory cytokines and chemokines, such as IL-1, IL-6, IL-8, or GM-CSF [105, 22, 31]. Combined with the pro-inflammatory cytokine production, TNF regulates osteoclastogenesis via several mechanisms [192]. TNF is a potent driver of osteoclast formation [142], but it also inhibits osteoblast differentiation and function [20], promoting the imbalance between bone destruction and formation as seen in RA [192]. However, this activity first described in whole bone depends on local cell interactions, and TNF promotes osteogenic differentiation of isolated human mesenchymal stem cell-derived osteoblasts, with a potent synergistic interaction with IL-17 [193]. The results of studies in several arthritis animal models also provide further evidence of the importance of TNF in RA [12]. For example, transgenic mice that express a modified human TNF gene spontaneously develop an inflammatory and destructive peripheral arthritis characterized by many hallmarks of RA (joint inflammation, bone erosion, and cartilage destruction). The treatment of these animals by a monoclonal anti-TNF antibody prevents disease development [134]. In the collagen-induced arthritis (CIA) model, the blockade of TNF by an anti-TNF antibody or by a soluble TNF-receptor fusion protein improves inflammation and joint damage [244, 245]. More importantly, the crucial role of TNF in RA has been confirmed by results from clinical trials in patients treated with TNF inhibitors [68, 67, 177]. Furthermore, clinical inhibition of TNF is associated with the suppression of neovascularization [194]. TNF blockade also reverses the increased nociception long before its effect on inflammation becomes obvious. Indeed, RA patients receiving an intravenous infusion of anti-TNF (infliximab) rapidly show a decreased central nervous system activity linked to nociceptive stimuli. This appears as early as 24 h after its administration, while the anti-inflammatory effects such as the improvement of joint swelling and DAS28 are detected later at 14 days [110]. In addition, the inhibition of TNF in RA patients promotes the emergence of a regulatory T cell population that could suppress effector T cells [178] and also reduce the leukocyte trafficking to the joints [229].
Treatments directed against TNF
Anti-TNF therapy is now fully part of RA treatment. Since 1999, three anti-TNF therapies have been approved: two monoclonal antibodies, infliximab (chimeric human/mouse) and adalimumab (human) and a fusion protein comprising human soluble TNFR linked to Fc component of human IgG1, etanercept. Since 2009, two other anti-TNF treatments have been authorized, certolizumab pegol and golimumab. Although all drugs share their clinical benefit, some differences between them have been observed in terms of rate of response, survival rate, tolerance, including the risk of TB [111, 135]. Currently, biosimilars, defined as highly similar to an already approved biological product, are starting to be approved and available: Inflectra and Remsima for infliximab, Erelzi for etanercept, and Amjevita for adalimumab.
Although anti-TNF treatments have provided a clear improvement in RA care, only two thirds of RA patients respond to anti-TNF leaving space for other treatments [111]. Furthermore, considering the biological role of TNF, adverse events, mainly an increased incidence of infections have been observed. Some studies have showed an increased incidence of some malignancies, but the direct effect of the TNF inhibitors remains controversial [204]. The use of TNF inhibitors is associated with an increased risk of TB reactivation [241]. As mentioned above, TNF plays a key role in the maintenance of granulomas, which is critical for protection from TB. Thus, the blockade of TNF promotes the deregulation of granulomas and the diffusion of TB infection. In addition, RA patients are more sensitive to infections notably to TB. Chronic inflammation induces a defect in the Th1 pathway, which is critical for protection from TB. Furthermore, IL-17 has an inhibitory effect on the Th1 pathway by inhibiting the expression of a functional IL-12 receptor [232]. Despite differences in the risk of TB reactivation between the different TNF inhibitors, this explains in part why the risk of reactivation is maximum at the beginning of TNF inhibition when the disease is still active. In addition, the use of TNF inhibitors has been associated with the development of autoimmunity. In most cases, this is limited to the induction of the production of autoantibodies [13].
IL-6
Signaling pathway and functions
Signaling pathway
IL-6 is a pleiotropic cytokine displaying very diverse activities and produced by almost all stromal and immune cells [120]. IL-6 exerts its biological effects through a fully competent IL-6 receptor (IL-6R), composed by two subunits: a type I cytokine α-receptor subunit (IL-6R, also known as CD126) and a common signal-transducing-β-receptor subunit (gp130, also known as CD130) [205] (Fig. 1). Fully active IL-6R expression is largely restricted to hepatocytes, leukocytes, and megakaryocytes, while gp130 expression is ubiquitous. The function of the IL-6 receptor requires the formation of an IL-6-IL-6R-gp130 complex. This complex is clustered into a dimer structure [217]. IL-6R displays two distinct modes of signaling, classical and trans-signaling.
The classical signaling is relevant only to cells that express the two IL-6R subunits, IL-6R and gp130, and it is mediated by the membrane-bound IL-6R subunit (mIL-6R) [127]. IL-6 binds first mIL-6R that leads to the homodimerization of gp130 and to a high-affinity functional receptor. In contrast, trans-signaling involves the soluble form of the IL-6R subunit (sIL-6R). This sIL-6R binds secreted IL-6 to form a complex displaying a higher circulating half-life than IL-6 alone and promoting its bioavailability [197]. The complex sIL-6R-IL-6 binds gp130 and thus could theoretically stimulate most cells, as gp130 is ubiquitously expressed. For example, synoviocytes express gp130 but not IL-6R and respond to IL-6 only in the presence of sIL-6R [171]. The sIL-6R subunit, which lacks the intracytoplasmic portion of mIL-6R, is produced by the enzymatic cleavage of mIL-6R or by alternative splicing. Furthermore, this trans-signaling is highly regulated by the soluble form of gp130 (sgp130), which is a natural inhibitor of IL-6 signaling. Indeed, exogenous sgp130 inhibits the binding of the complex sIL-6R-IL-6 to the mgp130 [181, 129].
In both mechanisms, the formation of the functional complex leads to signal transduction through gp130. Two pathways mediate this signal transduction, the Janus family tyrosine kinase-signal transducer and activator of transcription (JAK-STAT) pathway and the Ras-MAPK pathway. The negative regulation of IL-6 signaling is mediated by suppressor of cytokine signals (SOCS) [120].
Biological functions of IL-6
In health
IL-6 is an essential cytokine for the regulation of the immune system. In innate immunity, IL-6 modulates key functions including hematopoiesis, accumulation of neutrophils at infection sites through the control of granulopoiesis [151, 48]. In adaptive immunity, IL-6 is critical for immunoregulation. Furthermore, IL-6 plays a role in the regulation of T cells and also on B cells. IL-6 acts at a checkpoint in the differentiation of naïve T cells, promoting the pro-inflammatory Th17 cells and inhibiting the regulatory T cells. IL-6 is involved in the proliferation, survival commitment of T cells and in the modulation of their effector cytokine production [120]. IL-6 also controls the survival, expansion, and maturation of B cells and the production of IgG, IgM, and IgA [5, 120]. The biological role of IL-6 in innate and adaptive immunities is confirmed by an impaired response to viral, parasitic, and bacterial infections during IL-6 deficiency [141, 114, 185]. The central homeostatic processes and immunological outcomes controlled by IL-6 are mainly through the classical signaling [120].
In disease
If the classical signaling is more involved in health, IL-6 trans-signaling is important in disease. In different animal models (colitis, tissue fibrosis, inflammatory arthritis, neuroinflammation, etc.), the trans-signaling plays a key role in the recruitment and apoptosis of leukocytes, in the maintenance of effector T cells, and in the inflammatory activation of stromal tissues [126, 37]. For example, at sites of the disease, the CD4+ T cells lack the IL-6R subunit but they are still responsive to IL-6 through the trans-signaling pathway [125, 120]. Furthermore, in very different diseases, such as cardiac myxoma, a benign heart tumor, in RA, or in Castelman’s disease, an overproduction of IL-6 is detected, partly explaining the inflammatory symptoms [130, 112, 248]. Thus, IL-6 has a protective role in many acute situations such as infections, but its over-production in inflammatory diseases such as RA drives disease progression and chronicity [103].
IL-6 and RA
Involvement in the pathogenesis
IL-6 is a key cytokine in RA pathogenesis and mediates effects rather similar to those of TNF in the synovial environment. IL-6 is involved in various inflammatory effector pathways through activation of immune cells, endothelial cells, synoviocytes, or osteoclasts and production of acute-phase proteins such as CRP [103, 165]. In antigen-induced arthritis (AIA) and CIA, IL-6 plays a key role in the development of the disease [191, 6]. The deletion of IL-6 protects mice from the development of CIA, and the neutralization of IL-6 by an antibody improves the disease [6, 84]. In a monkey CIA model, the blockade of IL-6 by an antibody (tocilizumab (TCZ)) decreases joint swelling and the infiltration of inflammatory cells [234]. Furthermore, an overproduction of IL-6 and of sIL-6R is found in the synovial fluid and blood of RA patients and this excess correlates with disease activity and joint destruction [158, 206]. In cultured cells, IL-6 increases the production of chemokines by several cell types (endothelial cells, mononuclear cells, or synoviocytes) but also the expression of adhesion molecules [225]. This leads to an aggravation of the local inflammation by amplifying the inflammatory cell infiltrate. In the presence of sIL-6R, IL-6 increases the proliferation of synoviocytes and induces RANKL expression that promotes osteoclastogenesis [171, 102] and MMP (Matrix MetalloProteinases) production that contributes to cartilage degradation [226]. Moreover, the crucial proof of concept for a pivotal role for IL-6 in RA pathogenesis is provided by clinical trials in which the anti-IL-6R antibody TCZ suppresses disease activity and erosive progression in RA patients [219, 186]. In addition, first studies targeting the IL-6R demonstrate a significant decreased inflammation and clinical disease activity [187, 49]. Therefore, IL-6 is clearly a key cytokine in RA pathogenesis and thus a well-identified target for RA treatment.
Treatments involving IL-6 targeting
Currently, the anti-IL-6 treatment approved for RA is tocilizumab, which is a recombinant humanized anti-human IL-6R monoclonal antibody [8]. TCZ prevents IL-6 from binding to both mIL-6R and sIL-6R, thereby inhibiting the IL-6-mediated inflammatory effects [211]. In 2010, TCZ was first approved for the treatment of RA patients with active moderate-to-severe disease and an inadequate response to at least one disease-modifying antirheumatic drugs (DMARD) [211]. In 2012, TCZ was approved as a first-line biological agent. Adverse events include infections as for TNF inhibitors and also some more specific to IL-6 inhibition such as colon perforation or liver changes [211].
There are currently four other IL-6 inhibitors under development [165, 230, 227]. Sarilumab is a fully human monoclonal antibody against IL-6Rα, and it has been studied in phase IIb-III clinical trials [96, 119, 81]. Sirukumab is a humanized monoclonal anti-IL-6 antibody in phase II/III clinical trials [221] and phase III [144]. Olokizumab is a humanized anti-IL-6 monoclonal antibody that blocks the final assembly of the IL-6 signaling complex, and it has been investigated in a phase IIb study [95]. Clazakizumab is a humanized monoclonal antibody that potently binds IL-6 and has completed a phase IIb study [166, 242].
IL-1 and IL-1Ra
Signaling pathway and functions
Signaling pathway
IL-1, secreted mainly by macrophages, monocytes, and dendritic cells, was the first interleukin to be identified; it was described as a protein-inducing fever with two major proteins, IL-1α and IL-1β encoded by two distinct genes [64, 85, 90]. IL-1α and IL-1β are synthetized as precursor proteins, pro-IL-1α and pro-IL-1β. Pro-IL-1α is an already biologically active form while pro-IL-1β has no biological activity until it undergoes a proteolytic cleavage by the inflammasome-mediated caspase-1. IL-1α and IL-1β exert similar biological activities through the same receptor IL-1 type 1 receptor (IL-1R1) (Fig. 1). Activated by the ligand binding, the combination with the IL-1R accessory protein (IL-R1AcP) results in a fully active IL-1R1. IL-1R1 binds the adaptor protein myeloid differentiation primary response gene 88 (MyD88) that activates the protein kinase IL-1 receptor-associated kinase (IRAK4). Then IRAK4 phosphorylates and activates IRAK1 that is associated with TRAF6. This induces the oligomerization and the activation of TRAF6 that leads to the activation of the transcription factors NFκB and activator protein 1 (AP-1). In addition to IL-1R1, IL-1α and IL-1β can bind the IL-1 type II receptor (IL-1R2), which acts as a decoy receptor without signal transduction. Another important member of the IL-1 family is the IL-1 receptor antagonist (IL-1Ra). IL-1Ra can bind IL-1R1, but this binding inhibits the IL-1 signaling as IL-1Ra does not attract the IL-1R accessory protein-interacting domain, necessary for signal transduction. Thus, the effects of IL-1 are tightly controlled by the natural inhibitors IL-1R2 and IL-1Ra [85, 90].
Functions of IL-1
Health
IL-1 is a classic endogenous pyrogen, inducing fever, and was first identified as responsible for resistance to infections [237]. In addition, IL-1 modulates the differentiation and the function of innate and adaptive lymphoid cells [90, 215]. IL-1α is released upon cell necrosis and not actively secreted by cells [51]. This implies that IL-1α acts as an alarmin by rapidly mediating the early phases of sterile inflammation [47]. In contrast to necrosis where IL-1α triggers an inflammatory response by inducing pro-inflammatory cytokines such as IL-1β, IL-6, or TNF, during apoptosis IL-1α moves to the nucleus preventing the induction of inflammation [51]. If IL-1α is present in homeostatic conditions, IL-1β is produced upon inflammatory signals. IL-1β has a pro-inflammatory activity at tissue level, leading to vasodilation or activation of innate immune cells such as neutrophils. As such, it is an important component of the host defense against infections [184, 5]. IL-1β also acts on T cells, mainly by supporting the Th17 cell differentiation [2]. IL-1Ra is expressed in almost all tissues. Its major role is to prevent uncontrolled activation of IL-1R1 and thus IL-1β-mediated inflammation [90].
Disease
IL-1 is a key pro-inflammatory cytokine affecting a large target panel of cells like immune cells, endothelial cells, and of organs such as brain or liver. It is a major pathogenic factor of autoinflammation, autoimmunity, or infections. For example, an overproduction of IL-1β characterizes several genetically determined autoinflammatory disorders, such as Familial Mediterranean fever, associated with a mutation of the MEFV gene encoding an inflammasome component or as cryopyrin-associated periodic syndromes (CAPS) linked to a gain-of-function mutation in NLRP3 [222, 207].
At high concentrations, IL-1 is clearly associated with inflammatory pathogenesis and tumorigenesis. IL-1 plays a key role in chronic intestinal inflammation, as it promotes the production of many pro-inflammatory cytokines [63, 153]. Furthermore, high circulating levels of IL-1β are found in patients with infectious or inflammatory conditions [153]. IL-1β also promotes tumor angiogenesis and metastasis of tumors [38, 9]. IL-1 also plays a role in joint inflammation and cartilage degradation by activating osteoclastogenesis [124] and in pathogenesis of heart disease [26]. Thus, IL-1 is a pleiotropic pro-inflammatory cytokine involved in pathogenesis of several diseases, including RA.
IL-1 and RA
Involvement in the pathogenesis
IL-1 plays a major role in RA pathogenesis, mainly in bone and cartilage destruction, by stimulating the production of RANKL, involved in osteoclastogenesis and MMP, involved in cartilage degradation [59]. In RA joints, high levels of IL-1 (IL-1α and IL-1β) are detected in the synovial membrane and fluid [58, 235], with an imbalance between the levels of IL-1Ra and IL-1 [10]. Furthermore, several animal model studies demonstrate the importance of IL-1 in joint inflammation and subsequent tissue damage. The injection of IL-1α or IL-1β into the knee joints of rabbits or rats induces the accumulation of leukocytes in synovial fluid, cartilage degradation, and finally, chronic features of arthritis [107, 71, 46, 198]. The expression of human IL-1β following a gene transfer method leads to a severe and aggressive form of arthritis, similar to some features of human RA [98, 99]. In addition, IL-1 is also found in the inflamed synovium from mice with AIA and CIA [236, 86]. According to their background, IL-1Ra-deficient mice exhibit a more severe form of CIA [157] or develop spontaneously severe destructive arthritis [116]. The administration of IL-1Ra in immune complex-induced arthritis [238] and CIA [128] in mice and AIA in rabbits [11] limits cartilage and bone destruction. Thus, animal models provide a major evidence that IL-1 plays a key role in arthritis, mainly in tissue damage. In humans, several studies using an anti-IL-1 therapy (anakinra) demonstrate a significant but moderate improvement of clinical signs of RA [167, 52]. Anakinra exhibits a protective effect against bone and cartilage damage, but this effect is lower than in animal models.
Treatment
Anakinra is a recombinant IL-1R antagonist. In 2001, it was approved by the FDA for the treatment of moderate-to-severe RA with at least one failed DMARD therapy. Its overall clinical response is moderate and less potent than anti-TNF therapies in most patients [167, 33], possibly linked to its short half-life in plasma [52]. In contrast, IL-1 inhibition is highly effective in some auto-inflammatory diseases. Nevertheless, anakinra treatment has been shown to be safe, with no increased risk of infections in contrast to anti-TNF [25]. Another anti-IL-1 agent, canakinumab which is an anti-IL-1β antibody, has been investigated in a phase II study showing moderate improvement of signs and symptoms related to joint swelling [7]. Gevokizumab, an anti-IL-1β monoclonal antibody has also been investigated in a phase IIa study [91]. In addition, soluble type I IL-1 receptor has been tested with modest effect [65], and rilonacept, another IL-1 inhibitor, showed no effect in RA [162].
IL-17
Signaling pathway and functions
Signaling pathway
IL-17 is a pro-inflammatory cytokine mainly secreted by Th17 and other T cells and which exerts a major role in adaptive immunity. IL-17 binds and acts through a heterodimeric receptor composed of two chains, IL-17RA and IL-17RC [233]. The binding of IL-17 to IL-17RA induces the recruitment of IL-17RC that forms a complex (Fig. 1). Both chains contain a conserved signaling domain, the SEF/IL-17R (SEFIR) [189] which recruits the ubiquitin ligase Act1 through its own SEFIR [87, 203]. In turn, Act1, which also contains a TRAF-binding domain, recruits TRAF6 [150, 203] leading to the activation of the classical NFκB and the MAPK pathway and possibly the phosphoinositide-3 kinase (PI3K) pathway [88]. Such activation induces many inflammatory genes, mainly the neutrophil-specific chemokines [89, 250].
Functions of IL-17
Protective functions
IL-17 plays a key role in adaptive immunity, mainly in host defense against extra-cellular bacterial and fungal infections [140, 123, 176]. In these cases, the major function of IL-17 is neutrophil chemotaxis at the infection site which is critical for the microorganism clearance [121]. This is mediated by the induction of CXC chemokines, including IL-8 and growth factors such as GM-CSF [83]. The synergy of IL-17 with other pro-inflammatory cytokines, including TNF, IL-6, and IL-1, also promotes neutrophil activation. In addition to neutrophil recruitment, IL-17 induces the production of chemo-attractants for other immune cells such as lymphocytes, dendritic cells, or monocytes [176].
Pro-inflammatory functions
Although IL-17 has a crucial role in host defense, an excess of IL-17 is found in many chronic inflammatory and auto-immune diseases. This results in excessive pro-inflammatory cytokine and chemokine secretion, including TNF, IL-6, IL-1, and IL-8 by cells present at the inflammatory site notably monocytes and mesenchymal cells, leading to chronic inflammation. In addition, IL-17 stimulates the production of antimicrobial peptides required for skin protection, and MMP involved in tissue destruction [175, 42]. Thus, IL-17 contributes to the tissue destruction that occurs in chronic inflammatory diseases such as psoriasis, multiple sclerosis (MS), or RA. IL-17 also acts directly on phenotype and cell survival. In RA, IL-17, in synergy with TNF, increases the survival of synoviocytes and contributes to their aggressive and invasive phenotype [231, 117]. IL-17 contributes by different mechanisms to chronic inflammation, and thus, it is a key cytokine in several inflammatory diseases, notably in RA [17].
IL-17 and RA
Role of IL-17 in RA pathogenesis
Several studies in RA patients and in animal models demonstrate the major involvement of IL-17 in RA pathogenesis. IL-17 is spontaneously produced by RA synovium [40, 1, 249], and these increased IL-17 levels correlate with more severe joint damage and disease activity [137, 168]. Through synergy with other cytokines, such as TNF, IL-1, or IL-6, IL-17 leads to synoviocyte activation and cytokine production contributing to chronic inflammation [174, 17] and IL-17 also plays a role in neovascularization [115]. The addition of an anti-IL-17 in RA human synovium or bone explants reduces synovial inflammation and bone resorption [44]. In the CIA model, the intra-articular injection of IL-17 promotes arthritis with symptoms similar to human RA [43, 154] while in IL-17-deficient mice, the disease induction is suppressed [180]. Furthermore, the inhibition of IL-17 by a specific antibody decreases and slows down the arthritis progress [155, 138]. In the SKG model, the arthritis is inhibited in IL-17-deficient mice [113], and the administration of anti-IL-17 antibody decreases its progress [214]. In the streptococcal cell wall (SCW) model, the chronicity of the destructive synovitis requires the IL-17 pathway [156]. Thus, IL-17 participates to the induction and also to the progress of arthritis as a major factor in chronicity [17].
Targeting of IL-17
Secukinumab is a fully human monoclonal antibody against IL-17A. In 2010, a phase I clinical trial showed positive results in RA patients [118]. Current data from phase II trials are not fully convincing. Indeed, in a placebo-controlled phase II study, the difference between the two groups did not reach statistical significance [94]. Nevertheless, a long-term safety and efficacy evaluation in a 52-week follow-up showed an improvement and a sustained response to secukinumab [93]. More recently, various phase III studies have been completed or are still in progress [139]. The results are not as good as in psoriatic arthritis and ankylosing spondylitis where the drug is approved. Another anti-IL-17A monoclonal antibody ixekizumab has reached a similar conclusion [97]. In contrast, a monoclonal antibody directed against IL-17RA, brodalumab had no effect [195, 161], perhaps due to an inhibition of the anti-inflammatory cytokine IL-25, which also binds IL-17RA.
The situation might be more complex because the response to anti-IL-17 treatment displays a high heterogeneity between patients [118, 97, 28]. Some biomarkers such as the association with HLA-DRB1 alleles with clinical responses failed to predict the treatment response [28]. Thereby, there is a need of new biomarkers. A bioassay detecting circulating bioactive IL-17 could be used as a selection biomarker as not all RA patients have an IL-17-driven disease [183].
GM-CSF
Signaling pathway and functions
Signaling pathway
GM-CSF is a secreted cytokine, belonging to the colony-stimulating factor family of hematopoietic growth factors. GM-CSF binds the heterodimeric GM-CSF receptor (GM-CSFR) composed of a specific, low-affinity ligand-binding α-chain (GM-CSFRα), and a signal-transducing β-chain (GM-CSFRβ) which is analogous to gp130 discussed above [78, 101, 243]. The GM-CSF binding to GM-CSFRα induces the assembly of the signaling GM-CSFR complex (Fig. 1). GM-CSFR activation leads to the phosphorylation of JAK2 which is associated to GM-CSFRβ [101]. This initiates the JAK2/STAT5 signaling pathway [108]. The activation of JAK2 also leads to the phosphorylation of tyrosine residues on GM-CSFRβ that mediates the activation of other signaling pathways, including MAPK, PI3K-Akt, and NFκB [109, 108]. Src-like adaptor proteins negatively regulate GM-CSFR signaling by interacting with the GM-CSFRα [147]. SOCS also negatively regulate GM-CSFR signaling by targeting GM-CSFRβ for proteasome-mediated degradation [27].
Functions
Homeostatic functions
GM-CSF is produced by many cell types, including myeloid cells such as monocytes, immune cells such as lymphocytes, or tissue-resident cells such as fibroblasts [243]. GM-CSF is responsible for the proliferation and differentiation of myeloid cells from bone marrow progenitors [170]. Specifically, GM-CSF promotes bone marrow production of neutrophils, eosinophils, and monocytes; induces cell activation; and extends cell survival [170, 169, 57].
GM-CSF is also essential for the regulation of mature myeloid cell functions such as chemotaxis and cell adhesion, of dendritic cell functions, of pro-inflammatory cytokine production, and of phagocytosis [78, 54, 243]. It has a central role in regulating innate immunity notably by the polarization of macrophages into M1 pro-inflammatory phenotype [80, 160, 79, 212]. Thus, GM-CSF is not only a hematopoietic growth factor but is also involved in innate and adaptive immunities [239].
Pathologic functions
Patients with arthritic and inflammatory disorders have an increase of GM-CSF levels and GM-CSFR expression [146, 76, 35, 100, 75]. A high level of GM-CSF is also found in the serum of patients with acute aortic dissection or in the cerebrospinal fluid of patients with active MS [39]. In MS, GM-CSF also promotes the breakdown of the blood-brain barrier and the entry of inflammatory cells [201, 136]. Furthermore, GM-CSF acts as a pain mediator. GM-CSF activates the signal transduction in nociceptors, and leads to pain [209] in tumor and arthritis models, in a prostaglandin-dependent manner [55, 56]. In addition, GM-CSFR is expressed by sensory neurons, and GM-CSFR signaling sensitizes nociceptors in these neurons, leading to hyperalgesia and peripheral nerve remodeling [223].
GM-CSF and RA
Involvement in the pathogenesis
Different studies involve GM-CSF in RA pathogenesis. First, GM-CSF levels are increased in synovial fluid and blood from RA patients [246, 69, 16, 75], and GM-CSFRα is also up-regulated in inflamed synovial tissue and in circulating mononuclear cells [76, 18]. Moreover, the administration of GM-CSF to treat neutropenia in RA patients has been followed by disease flare [196, 106, 60]. In animal models of arthritis, while deficiency or blockade of GM-CSF is protective, the overexpression or injection of GM-CSF promote disease. GM-CSF-deficient mice are protected from CIA development [36] or from methylated BSA/IL-1-induced arthritis [143]. In addition, the blockade of GM-CSF improves arthritis in the CIA model [53], mBSA/IL-1-induced arthritis [247], or in SKG models [214] and reduces inflammation and cartilage degradation in the SCW model [199]. Conversely, the administration of GM-CSF exacerbates arthritis in the CIA model [34]. Furthermore, as GM-CSF acts as a pain mediator, the high levels found in inflammatory synovium could be linked to joint pain, and GM-CSF-deficient mice are protected from this type of pain [55, 56].
Targeting of GM-CSF
Mavrilimumab is a fully human antibody that binds GM-CSFRα and thus inhibits GM-CSF signaling. It has been successfully tested in phase I/II trials with no serious adverse events [29, 30]. Another fully human antibody targeting GM-CSF itself, MOR103, has shown preliminary evidences of efficacy in a phase Ib/IIa clinical trial [15, 239]. Early studies on three other anti-GM-CSF are currently ongoing with lenzilumab, namilumab, and MORAb-022 in phase I [213]. Despite rapid treatment responses to mavrilimumab and MOR103 which provide evidence of clinical efficacy, further clinical investigation is necessary to determine the position of anti-GM-CSF therapy specifically in patients who did not respond to other available DMARDs.
Cytokine interactions and regulation
Interactions between cytokines
During RA pathogenesis, cytokines clearly play a central role. In addition to their own specific action, they act in synergy to increase the inflammatory processes and to maintain their own production (Fig. 2). These cytokines also interact with each other at the production level, as an example, IL-17 increasing IL-1 and TNF production by monocytes [174]. IL-17, IL-1, and TNF stimulate stromal cells to produce IL-6, and the combination of these cytokines leads to synergistic or additive effects [83, 41]. This synergistic effect is also extended to chemokine production such as CCL20, which in turn attracts Th17 cells [45]. The synergy between IL-1 and TNF induces the expression of endothelial adhesion molecules or GM-CSF by synovial cells [243]. Synergistic effects between IL-17, TNF, and IL-1 have been demonstrated in many cell types like synoviocytes, chondrocytes, osteoblasts, or myoblasts [174]. Furthermore, a complementary action of GM-CSF and IL-17 leads to a more severe pathology in experimental arthritis [240] and also further stimulates granulopoiesis and dendritic cell expansion [149]. Thereby, the key cytokines involved in RA pathogenesis, act often in synergy to increase and maintain the inflammation. This synergy could lead to new therapeutics by combining cytokine blockade [200, 19].

Interactions between cells and cytokines during RA pathogenesis. During RA, immune cells migrate to the inflammatory site, where they interact with local synoviocytes. This interaction leads to the production of cytokines that in turn activate immune and mesenchymal cells and promote their own secretion. For example, monocytes secrete IL-6 and IL-1β which promote Th17 differentiation. Th17 cells secrete IL-17 which contributes to chronicity, activating synoviocytes to produce IL-6 and monocytes to produce IL-1 and TNF. IL-1 and TNF act in synergy on synoviocytes to produce IL-6 and GM-CSF and synergize with IL-17 increasing IL-6 production. Synoviocytes secrete IL-6, which exerts a positive feedback contributing to hyperplasia. IL-6 also activates Th17 cells to produce IL-17. In addition, IL-6 acts on osteoblasts to produce RANKL which stimulates osteoclasts, promoting bone destruction. Osteoclasts are also activated by IL-1 and TNF. The stimulation of synoviocytes by the different cytokines leads to the production of chemokines, which recruit leukocytes amplifying the inflammatory response, and the production of MMP promoting cartilage damage. Thereby, cell interactions and cytokines themselves regulate cytokine production and contribute to the pro-inflammatory environment and its maintenance
Regulation of cytokine production
As described above, cytokines act in synergy and have a regulatory effect on their production. Furthermore, another key point of regulation at the local site is the cell-cell interactions. During RA, immune cells migrate to the inflammatory site in the synovium and interact with local mesenchymal cells, synoviocytes, which then modulate the synovial cytokine profile [14]. These interactions alone lead to the pro-inflammatory cytokine production, notably IL-6 and IL-1β. The cell contact between activated immune cells and synoviocytes also leads to a high IL-17 secretion that could explain the presence of IL-17 in patient joints [188]. Thereby, cellular interactions are a key point in the induction and maintenance of massive cytokine production.
Another connecting point for cytokines is the JAK/STAT pathway. The activation of STAT proteins by cytokine-receptor binding results in JAK phosphorylation. This pathway is a conserved signaling pathway used by many cytokines, including IL-6 and GM-CSF [159, 190]. In RA patients, abnormalities in JAK/STAT pathway have been observed which correlate with high levels of cytokines, such as IL-6 [122]. Thereby, this pathway has become an attractive therapeutic target. Currently, tofacitinib, a small molecule JAK1/3 inhibitor has been approved by the FDA for the treatment of moderate-to-severe RA and several other JAK inhibitors are undergoing phase II trials [190]. These new therapeutics could represent a valuable addition to the current DMARDs and biologic agents (Fig. 3).

Current and future agents in RA targeting cytokines. Biologic agents target cytokines or their receptors. Infliximab, etanercept, adalimumab, certolizumab, and golimumab bind TNF, thereby inhibiting TNF-TNFR binding. Canakinumab and gevokizumab are monoclonal antibodies under development, and both bind IL-1β. Anakinra inhibits also IL-1 signaling by increased IL-1R occupancy without signaling. IL-17 is targeted by two antibodies under development, secukinumab and ixekizumab. Brodalumab targets IL-17RA and prevents IL-17 binding. Several biologic agents targeting GM-CSF itself are under development: MOR103, lenzilumab, namilumab, and MORAb-022. Mavrilimumab inhibits GM-CSF signaling by acting on GM-CSFRα. The only approved biologic agent inhibiting IL-6 signaling is tocilizumab which binds to mIL-6R and sIL-6R. Sarilumab, under development, also targets IL-6R. Three antibodies targeting directly IL-6 are under development, sirukumab, olokizumab, and clazakizumab. A new generation of small molecules targeting JAK/STAT pathway are under development. One of them, tofacitinib, which inhibits JAK, is now approved in some countries
Unresolved aspects
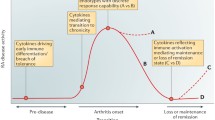
The induction of RA remission is now the therapeutic objective. Despite the improvement of clinical symptoms and joint damage, molecular and cellular heterogeneity between patients impacts the response to therapies. Cytokines are involved in each phase of RA pathogenesis by contributing to autoimmunity, chronic inflammation, and tissue damage [164]. Nevertheless, choosing the right cytokine target for a given patient with a given disease is not easy as their hierarchical relationship is not yet clear. A linear model is proposed where TNF would drive downstream cytokines, including IL-6 or IL-1. This model seems to explain the dominance of TNF in synovial culture systems and thus the dominance of anti-TNF treatments [73]. Although biologic therapies have improved the management and outcome, the patients who achieve disease remission remain a minority [210, 133] and the biological DMARDs induce more adverse events, specifically infections than conventional synthetic DMARDs [204, 148].
Therefore, new strategies to reach higher rates of response and better safety with an optimum risk benefit are needed. In this way, combination therapy or bispecific molecules are options. Indeed, based on their synergistic interactions, bispecific molecules against TNF and IL-17 are under development [19]. The combination therapy remains to be clarified as the first studies showed failures, as for the combination of etanercept and anakinra [92]. Another way is to identify predictive biomarkers of response and tolerance. For example, the presence of autoantibodies against pro-inflammatory cytokines, such as IL-1α or more recently IL-17 has been described as a marker for a good prognostic in RA [131, 173, 182]. In addition, the synovial phenotype could correlate with the response to biologic therapies [62]. Such advance could allow a stratification of patients according to their disease and lead to a personalized treatment for each patient. Several studies have focused on genetic factors associated with treatment response [228]. For example, several single nucleotide polymorphisms in the promoter region of TNF are related to clinical responses to treatment [228, 61, 82]. At this stage, however, these markers are not yet established companion biomarkers.
Conclusion
Targeting of cytokines has been a major progress in RA treatment. Despite such advances made over the last years, many issues remain open. The use of new powerful tools and increased knowledge may lead to a better understanding of RA pathogenesis to select appropriate treatments for each patient and reach remission. Furthermore, little is known about RA patients who achieve a state of remission. Some evidence suggests that early and aggressive treatment is an important factor to reach remission [4, 179]. Most patients who achieve a remission state display less deterioration of function and radiographic progression. Nevertheless, some of them still have radiographic progression despite persistent clinical remission. Thereby, understanding the mechanisms of remission as well of chronicity may provide new means to reach a final cure.
Abbreviations
- RA:
-
Rheumatoid arthritis
- TNF(R):
-
Tumor necrosis factor (receptor)
- IL:
-
Interleukin
- TRADD:
-
TNFR1-associated death domain protein
- NFκB:
-
Nuclear factor κB
- MAPKs:
-
Mitogen-activated protein kinases
- MLKL:
-
Mixed lineage kinase domain-like protein
- TRAF:
-
TNFR-associated factor
- PKB:
-
protein kinase B
- TB:
-
Mycobacterium tuberculosis
- CIA:
-
Collagen-induced arthritis
- AIA:
-
Antigen-induced arthritis
- FDA:
-
Food and Drug Administration
- TCZ:
-
Tocilizumab
- JAK:
-
Janus family tyrosine kinase
- STAT:
-
Signal transducer and activator of transcription
- SOCS:
-
Suppressor of cytokine signals
- MyD88:
-
Myeloid differentiation primary response gene 88
- IRAK:
-
IL-1 receptor-associated kinase
- AP-1:
-
Activator protein 1
- PI3K:
-
Phosphoinositide-3 kinase
- CAPS:
-
Cryopyrin-associated periodic syndromes
- MS:
-
Multiple sclerosis
- DMARD:
-
Disease-modifying antirheumatic drugs
References
Aarvak T, Chabaud M, Miossec P, Natvig JB (1999) IL-17 is produced by some proinflammatory Th1/Th0 cells but not by Th2 cells. J Immunol 162:1246–1251
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8:942–949
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat Rev Immunol 3:745–756
Ajeganova S, van Steenbergen HW, van Nies JA, Burgers LE, Huizinga TW, van der Helm-van Mil AH (2015) Disease-modifying antirheumatic drug-free sustained remission in rheumatoid arthritis: an increasingly achievable outcome with subsidence of disease symptoms. Ann Rheum Dis 75:867–873
Akdis M, Aab A, Altunbulakli C, Azkur K, Costa RA, Crameri R, Duan S, Eiwegger T, Eljaszewicz A, Ferstl R, Frei R, Garbani M, Globinska A, Hess L, Huitema C, Kubo T, Komlosi Z, Konieczna P, Kovacs N, Kucuksezer UC, Meyer N, Morita H, Olzhausen J, O’Mahony L, Pezer M, Prati M, Rebane A, Rhyner C, Rinaldi A, Sokolowska M, Stanic B, Sugita K, Treis A, van de Veen W, Wanke K, Wawrzyniak M, Wawrzyniak P, Wirz OF, Zakzuk JS, Akdis CA (2016) Interleukins (from IL-1 to IL-38), interferons, transforming growth factor beta, and TNF-alpha: Receptors, functions, and roles in diseases. J Allergy Clin Immunol 138:984–1010
Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G (1998) Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 187:461–468
Alten R, Gomez-Reino J, Durez P, Beaulieu A, Sebba A, Krammer G, Preiss R, Arulmani U, Widmer A, Gitton X, Kellner H (2011) Efficacy and safety of the human anti-IL-1beta monoclonal antibody canakinumab in rheumatoid arthritis: results of a 12-week, phase II, dose-finding study. BMC Musculoskelet Disord 12:153
Alves JD, Marinho A, Serra MJ (2011) Tocilizumab: is there life beyond anti-TNF blockade? Int J Clin Pract 65:508–513
Apte RN, Dotan S, Elkabets M, White MR, Reich E, Carmi Y, Song X, Dvozkin T, Krelin Y, Voronov E (2006) The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev 25:387–408
Arend WP (2001) Cytokine imbalance in the pathogenesis of rheumatoid arthritis: the role of interleukin-1 receptor antagonist. Semin Arthritis Rheum 30:1–6
Arner EC, Harris RR, DiMeo TM, Collins RC, Galbraith W (1995) Interleukin-1 receptor antagonist inhibits proteoglycan breakdown in antigen induced but not polycation induced arthritis in the rabbit. J Rheumatol 22:1338–1346
Asquith DL, Miller AM, McInnes IB, Liew FY (2009) Animal models of rheumatoid arthritis. Eur J Immunol 39:2040–2044
Atzeni F, Talotta R, Salaffi F, Cassinotti A, Varisco V, Battellino M, Ardizzone S, Pace F, Sarzi-Puttini P (2013) Immunogenicity and autoimmunity during anti-TNF therapy. Autoimmun Rev 12:703–708
Bartok B, Firestein GS (2010) Fibroblast-like synoviocytes: key effector cells in rheumatoid arthritis. Immunol Rev 233:233–255
Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, Berenfus VY, Vladeva S, Rech J, Rubbert-Roth A, Korkosz M, Rekalov D, Zupanets IA, Ejbjerg BJ, Geiseler J, Fresenius J, Korolkiewicz RP, Schottelius AJ, Burkhardt H (2014) MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis 74:1058–1064
Bell AL, Magill MK, McKane WR, Kirk F, Irvine AE (1995) Measurement of colony-stimulating factors in synovial fluid: potential clinical value. Rheumatol Int 14:177–182
Benedetti G, Miossec P (2014) Interleukin 17 contributes to the chronicity of inflammatory diseases such as rheumatoid arthritis. Eur J Immunol 44:339–347
Berenbaum F, Rajzbaum G, Amor B, Toubert A (1994) Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw 5:43–46
Beringer A, Noack M, Miossec P (2016) IL-17 in chronic inflammation: from discovery to targeting. Trends Mol Med 22:230–241
Bertolini DR, Nedwin GE, Bringman TS, Smith DD, Mundy GR (1986) Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 319:516–518
Braumuller H, Wieder T, Brenner E, Assmann S, Hahn M, Alkhaled M, Schilbach K, Essmann F, Kneilling M, Griessinger C, Ranta F, Ullrich S, Mocikat R, Braungart K, Mehra T, Fehrenbacher B, Berdel J, Niessner H, Meier F, van den Broek M, Haring HU, Handgretinger R, Quintanilla-Martinez L, Fend F, Pesic M, Bauer J, Zender L, Schaller M, Schulze-Osthoff K, Rocken M (2013) T-helper-1-cell cytokines drive cancer into senescence. Nature 494:361–365
Brennan FM, Chantry D, Jackson A, Maini R, Feldmann M (1989) Inhibitory effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid arthritis. Lancet 2:244–247
Brennan FM, McInnes IB (2008) Evidence that cytokines play a role in rheumatoid arthritis. J Clin Invest 118:3537–3545
Brenner D, Blaser H, Mak TW (2015) Regulation of tumour necrosis factor signalling: live or let die. Nat Rev Immunol 15:362–374
Bresnihan B (2001) The safety and efficacy of interleukin-1 receptor antagonist in the treatment of rheumatoid arthritis. Semin Arthritis Rheum 30:17–20
Bujak M, Frangogiannis NG (2009) The role of IL-1 in the pathogenesis of heart disease. Arch Immunol Ther Exp 57:165–176
Bunda S, Kommaraju K, Heir P, Ohh M (2013) SOCS-1 mediates ubiquitylation and degradation of GM-CSF receptor. PLoS One 8:e76370
Burmester GR, Durez P, Shestakova G, Genovese MC, Schulze-Koops H, Li Y, Wang YA, Lewitzky S, Koroleva I, Berneis AA, Lee DM, Hueber W (2016) Association of HLA-DRB1 alleles with clinical responses to the anti-interleukin-17A monoclonal antibody secukinumab in active rheumatoid arthritis. Rheumatology (Oxford) 55:49–55
Burmester GR, Feist E, Sleeman MA, Wang B, White B, Magrini F (2011) Mavrilimumab, a human monoclonal antibody targeting GM-CSF receptor-alpha, in subjects with rheumatoid arthritis: a randomised, double-blind, placebo-controlled, phase I, first-in-human study. Ann Rheum Dis 70:1542–1549
Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, Szombati I, Esfandiari E, Sleeman MA, Kane CD, Cavet G, Wang B, Godwood A, Magrini F (2012) Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann Rheum Dis 72:1445–1452
Butler DM, Maini RN, Feldmann M, Brennan FM (1995) Modulation of proinflammatory cytokine release in rheumatoid synovial membrane cell cultures. Comparison of monoclonal anti TNF-alpha antibody with the interleukin-1 receptor antagonist. Eur Cytokine Netw 6:225–230
Bystrom J, Clanchy FI, Taher TE, Mangat P, Jawad AS, Williams RO, Mageed RA (2016) TNFalpha in the regulation of Treg and Th17 cells in rheumatoid arthritis and other autoimmune inflammatory diseases. Cytokine
Callhoff J, Weiss A, Zink A, Listing J (2013) Impact of biologic therapy on functional status in patients with rheumatoid arthritis—a meta-analysis. Rheumatology (Oxford) 52:2127–2135
Campbell IK, Bendele A, Smith DA, Hamilton JA (1997) Granulocyte-macrophage colony stimulating factor exacerbates collagen induced arthritis in mice. Ann Rheum Dis 56:364–368
Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA (1991) Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol 147:1238–1246
Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA (1998) Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol 161:3639–3644
Campbell IL, Erta M, Lim SL, Frausto R, May U, Rose-John S, Scheller J, Hidalgo J (2014) Trans-signaling is a dominant mechanism for the pathogenic actions of interleukin-6 in the brain. J Neurosci 34:2503–2513
Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, Abutbul S, Huszar M, Dinarello CA, Apte RN, Voronov E (2013) The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol 190:3500–3509
Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O (1998) Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol 20:373–382
Chabaud M, Durand JM, Buchs N, Fossiez F, Page G, Frappart L, Miossec P (1999) Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum 42:963–970
Chabaud M, Fossiez F, Taupin JL, Miossec P (1998) Enhancing effect of IL-17 on IL-1-induced IL-6 and leukemia inhibitory factor production by rheumatoid arthritis synoviocytes and its regulation by Th2 cytokines. J Immunol 161:409–414
Chabaud M, Garnero P, Dayer JM, Guerne PA, Fossiez F, Miossec P (2000) Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine 12:1092–1099
Chabaud M, Lubberts E, Joosten L, van Den Berg W, Miossec P (2001) IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res 3:168–177
Chabaud M, Miossec P (2001) The combination of tumor necrosis factor alpha blockade with interleukin-1 and interleukin-17 blockade is more effective for controlling synovial inflammation and bone resorption in an ex vivo model. Arthritis Rheum 44:1293–1303
Chabaud M, Page G, Miossec P (2001) Enhancing effect of IL-1, IL-17, and TNF-alpha on macrophage inflammatory protein-3alpha production in rheumatoid arthritis: regulation by soluble receptors and Th2 cytokines. J Immunol 167:6015–6020
Chandrasekhar S, Harvey AK, Hrubey PS, Bendele AM (1990) Arthritis induced by interleukin-1 is dependent on the site and frequency of intraarticular injection. Clin Immunol Immunopathol 55:382–400
Chen CJ, Kono H, Golenbock D, Reed G, Akira S, Rock KL (2007) Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13:851–856
Chou DB, Sworder B, Bouladoux N, Roy CN, Uchida AM, Grigg M, Robey PG, Belkaid Y (2012) Stromal-derived IL-6 alters the balance of myeloerythroid progenitors during Toxoplasma gondii infection. J Leukoc Biol 92:123–131
Choy EH, Isenberg DA, Garrood T, Farrow S, Ioannou Y, Bird H, Cheung N, Williams B, Hazleman B, Price R, Yoshizaki K, Nishimoto N, Kishimoto T, Panayi GS (2002) Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: a randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum 46:3143–3150
Choy EH, Kavanaugh AF, Jones SA (2013) The problem of choice: current biologic agents and future prospects in RA. Nat Rev Rheumatol 9:154–163
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN (2010) Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A 107:2574–2579
Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, Kremer J, Bear MB, Rich WJ, McCabe D (2002) Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46:614–624
Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA (2001) Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res 3:293–298
Cook AD, Braine EL, Hamilton JA (2004) Stimulus-dependent requirement for granulocyte-macrophage colony-stimulating factor in inflammation. J Immunol 173:4643–4651
Cook AD, Pobjoy J, Sarros S, Steidl S, Durr M, Lacey DC, Hamilton JA (2013) Granulocyte-macrophage colony-stimulating factor is a key mediator in inflammatory and arthritic pain. Ann Rheum Dis 72:265–270
Cook AD, Pobjoy J, Steidl S, Durr M, Braine EL, Turner AL, Lacey DC, Hamilton JA (2012) Granulocyte-macrophage colony-stimulating factor is a key mediator in experimental osteoarthritis pain and disease development. Arthritis Res Ther 14:R199
Cowburn AS, Summers C, Dunmore BJ, Farahi N, Hayhoe RP, Print CG, Cook SJ, Chilvers ER (2011) Granulocyte/macrophage colony-stimulating factor causes a paradoxical increase in the BH3-only pro-apoptotic protein Bim in human neutrophils. Am J Respir Cell Mol Biol 44:879–887
Dayer JM, Bresnihan B (2002) Targeting interleukin-1 in the treatment of rheumatoid arthritis. Arthritis Rheum 46:574–578
Dayer JM, de Rochemonteix B, Burrus B, Demczuk S, Dinarello CA (1986) Human recombinant interleukin 1 stimulates collagenase and prostaglandin E2 production by human synovial cells. J Clin Invest 77:645–648
de Vries EG, Willemse PH, Biesma B, Stern AC, Limburg PC, Vellenga E (1991) Flare-up of rheumatoid arthritis during GM-CSF treatment after chemotherapy. Lancet 338:517–518
de Vries N, Tak PP (2005) The response to anti-TNF-alpha treatment: gene regulation at the bedside. Rheumatology (Oxford) 44:705–707
Dennis G Jr, Holweg CT, Kummerfeld SK, Choy DF, Setiadi AF, Hackney JA, Haverty PM, Gilbert H, Lin WY, Diehl L, Fischer S, Song A, Musselman D, Klearman M, Gabay C, Kavanaugh A, Endres J, Fox DA, Martin F, Townsend MJ (2014) Synovial phenotypes in rheumatoid arthritis correlate with response to biologic therapeutics. Arthritis Res Ther 16:R90
Dinarello CA (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117:3720–3732
Dinarello CA, Renfer L, Wolff SM (1977) Human leukocytic pyrogen: purification and development of a radioimmunoassay. Proc Natl Acad Sci U S A 74:4624–4627
Drevlow BE, Lovis R, Haag MA, Sinacore JM, Jacobs C, Blosche C, Landay A, Moreland LW, Pope RM (1996) Recombinant human interleukin-1 receptor type I in the treatment of patients with active rheumatoid arthritis. Arthritis Rheum 39:257–265
Elinav E, Nowarski R, Thaiss CA, Hu B, Jin C, Flavell RA (2013) Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat Rev Cancer 13:759–771
Elliott MJ, Maini RN, Feldmann M, Kalden JR, Antoni C, Smolen JS, Leeb B, Breedveld FC, Macfarlane JD, Bijl H et al (1994) Randomised double-blind comparison of chimeric monoclonal antibody to tumour necrosis factor alpha (cA2) versus placebo in rheumatoid arthritis. Lancet 344:1105–1110
Elliott MJ, Maini RN, Feldmann M, Long-Fox A, Charles P, Katsikis P, Brennan FM, Walker J, Bijl H, Ghrayeb J et al (1993) Treatment of rheumatoid arthritis with chimeric monoclonal antibodies to tumor necrosis factor alpha. Arthritis Rheum 36:1681–1690
Farahat MN, Yanni G, Poston R, Panayi GS (1993) Cytokine expression in synovial membranes of patients with rheumatoid arthritis and osteoarthritis. Ann Rheum Dis 52:870–875
Faustman DL, Davis M (2013) TNF receptor 2 and disease: autoimmunity and regenerative medicine. Front Immunol 4:478
Feige U, Karbowski A, Rordorf-Adam C, Pataki A (1989) Arthritis induced by continuous infusion of hr-interleukin-1 alpha into the rabbit knee-joint. Int J Tissue React 11:225–238
Feldmann M (2009) Translating molecular insights in autoimmunity into effective therapy. Annu Rev Immunol 27:1–27
Feldmann M, Brennan FM, Maini RN (1996) Rheumatoid arthritis. Cell 85:307–310
Feldmann M, Brennan FM, Maini RN (1996) Role of cytokines in rheumatoid arthritis. Annu Rev Immunol 14:397–440
Fiehn C, Wermann M, Pezzutto A, Hufner M, Heilig B (1992) Plasma GM-CSF concentrations in rheumatoid arthritis, systemic lupus erythematosus and spondyloarthropathy. Z Rheumatol 51:121–126
Field M, Clinton L (1993) Expression of GM-CSF receptor in rheumatoid arthritis. Lancet 342:1244
Firestein GS (2014) The disease formerly known as rheumatoid arthritis. Arthritis Res Ther 16:114
Fleetwood AJ, Cook AD, Hamilton JA (2005) Functions of granulocyte-macrophage colony-stimulating factor. Crit Rev Immunol 25:405–428
Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA (2009) GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol 86:411–421
Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD (2007) Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol 178:5245–5252
Fleischmann R, van Adelsberg J, Lin Y, da Rocha Castelar-Pinheiro G, Brzezicki J, Hrycaj P, Graham NM, van Hoogstraten H, Bauer D, Burmester GR (2016) Sarilumab and non-biologic disease-modifying antirheumatic drugs in patients with active RA and inadequate response or intolerance to TNF inhibitors. Arthritis Rheumatol
Fonseca JE, Carvalho T, Cruz M, Nero P, Sobral M, Mourao AF, Cavaleiro J, Ligeiro D, Abreu I, Carmo-Fonseca M, Branco JC (2005) Polymorphism at position −308 of the tumour necrosis factor alpha gene and rheumatoid arthritis pharmacogenetics. Ann Rheum Dis 64:793–794
Fossiez F, Djossou O, Chomarat P, Flores-Romo L, Ait-Yahia S, Maat C, Pin JJ, Garrone P, Garcia E, Saeland S, Blanchard D, Gaillard C, Das Mahapatra B, Rouvier E, Golstein P, Banchereau J, Lebecque S (1996) T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med 183:2593–2603
Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, Ohsugi Y, Nishikawa T, Ripley B, Kimura A, Kishimoto T, Naka T (2008) Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 58:3710–3719
Gabay C, Lamacchia C, Palmer G (2010) IL-1 pathways in inflammation and human diseases. Nat Rev Rheumatol 6:232–241
Gabay C, Marinova-Mutafchieva L, Williams RO, Gigley JP, Butler DM, Feldmann M, Arend WP (2001) Increased production of intracellular interleukin-1 receptor antagonist type I in the synovium of mice with collagen-induced arthritis: a possible role in the resolution of arthritis. Arthritis Rheum 44:451–462
Gaffen SL (2011) Recent advances in the IL-17 cytokine family. Curr Opin Immunol 23:613–619
Gaffen SL (2009) Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 9:556–567
Gaffen SL, Jain R, Garg AV, Cua DJ (2014) The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol 14:585–600
Garlanda C, Dinarello CA, Mantovani A (2013) The interleukin-1 family: back to the future. Immunity 39:1003–1018
Geiler J, McDermott MF (2010) Gevokizumab, an anti-IL-1beta mAb for the potential treatment of type 1 and 2 diabetes, rheumatoid arthritis and cardiovascular disease. Curr Opin Mol Ther 12:755–769
Genovese MC, Cohen S, Moreland L, Lium D, Robbins S, Newmark R, Bekker P (2004) Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum 50:1412–1419
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, Hugot S, Ligozio G, Mpofu S (2014) One-year efficacy and safety results of secukinumab in patients with rheumatoid arthritis: phase II, dose-finding, double-blind, randomized, placebo-controlled study. J Rheumatol 41:414–421
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, Hugot S, Mpofu S (2013) Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis 72:863–869
Genovese MC, Fleischmann R, Furst D, Janssen N, Carter J, Dasgupta B, Bryson J, Duncan B, Zhu W, Pitzalis C, Durez P, Kretsos K (2014) Efficacy and safety of olokizumab in patients with rheumatoid arthritis with an inadequate response to TNF inhibitor therapy: outcomes of a randomised phase IIb study. Ann Rheum Dis 73:1607–1615
Genovese MC, Fleischmann R, Kivitz AJ, Rell-Bakalarska M, Martincova R, Fiore S, Rohane P, van Hoogstraten H, Garg A, Fan C, van Adelsberg J, Weinstein SP, Graham NM, Stahl N, Yancopoulos GD, Huizinga TW, van der Heijde D (2015) Sarilumab plus methotrexate in patients with active rheumatoid arthritis and inadequate response to methotrexate: results of a phase III Study. Arthritis Rheumatol 67:1424–1437
Genovese MC, Greenwald M, Cho CS, Berman A, Jin L, Cameron GS, Benichou O, Xie L, Braun D, Berclaz PY, Banerjee S (2014) A phase II randomized study of subcutaneous ixekizumab, an anti-interleukin-17 monoclonal antibody, in rheumatoid arthritis patients who were naive to biologic agents or had an inadequate response to tumor necrosis factor inhibitors. Arthritis Rheumatol 66:1693–1704
Ghivizzani SC, Kang R, Georgescu HI, Lechman ER, Jaffurs D, Engle JM, Watkins SC, Tindal MH, Suchanek MK, McKenzie LR, Evans CH, Robbins PD (1997) Constitutive intra-articular expression of human IL-1 beta following gene transfer to rabbit synovium produces all major pathologies of human rheumatoid arthritis. J Immunol 159:3604–3612
Ghivizzani SC, Lechman ER, Tio C, Mule KM, Chada S, McCormack JE, Evans CH, Robbins PD (1997) Direct retrovirus-mediated gene transfer to the synovium of the rabbit knee: implications for arthritis gene therapy. Gene Ther 4:977–982
Hamilton JA (2008) Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 8:533–544
Hansen G, Hercus TR, McClure BJ, Stomski FC, Dottore M, Powell J, Ramshaw H, Woodcock JM, Xu Y, Guthridge M, McKinstry WJ, Lopez AF, Parker MW (2008) The structure of the GM-CSF receptor complex reveals a distinct mode of cytokine receptor activation. Cell 134:496–507
Hashizume M, Hayakawa N, Mihara M (2008) IL-6 trans-signalling directly induces RANKL on fibroblast-like synovial cells and is involved in RANKL induction by TNF-alpha and IL-17. Rheumatology (Oxford) 47:1635–1640
Hashizume M, Mihara M (2011) The roles of interleukin-6 in the pathogenesis of rheumatoid arthritis. Arthritis 2011:765624
Haudek SB, Taffet GE, Schneider MD, Mann DL (2007) TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest 117:2692–2701
Haworth C, Brennan FM, Chantry D, Turner M, Maini RN, Feldmann M (1991) Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: regulation by tumor necrosis factor-alpha. Eur J Immunol 21:2575–2579
Hazenberg BP, Van Leeuwen MA, Van Rijswijk MH, Stern AC, Vellenga E (1989) Correction of granulocytopenia in Felty’s syndrome by granulocyte-macrophage colony-stimulating factor. Simultaneous induction of interleukin-6 release and flare-up of the arthritis. Blood 74:2769–2770
Henderson B, Pettipher ER (1988) Comparison of the in vivo inflammatory activities after intra-articular injection of natural and recombinant IL-1 alpha and IL-1 beta in the rabbit. Biochem Pharmacol 37:4171–4176
Hercus TR, Dhagat U, Kan WL, Broughton SE, Nero TL, Perugini M, Sandow JJ, D’Andrea RJ, Ekert PG, Hughes T, Parker MW, Lopez AF (2013) Signalling by the betac family of cytokines. Cytokine Growth Factor Rev 24:189–201
Hercus TR, Thomas D, Guthridge MA, Ekert PG, King-Scott J, Parker MW, Lopez AF (2009) The granulocyte-macrophage colony-stimulating factor receptor: linking its structure to cell signaling and its role in disease. Blood 114:1289–1298
Hess A, Axmann R, Rech J, Finzel S, Heindl C, Kreitz S, Sergeeva M, Saake M, Garcia M, Kollias G, Straub RH, Sporns O, Doerfler A, Brune K, Schett G (2011) Blockade of TNF-alpha rapidly inhibits pain responses in the central nervous system. Proc Natl Acad Sci U S A 108:3731–3736
Hetland ML, Christensen IJ, Tarp U, Dreyer L, Hansen A, Hansen IT, Kollerup G, Linde L, Lindegaard HM, Poulsen UE, Schlemmer A, Jensen DV, Jensen S, Hostenkamp G, Ostergaard M (2010) Direct comparison of treatment responses, remission rates, and drug adherence in patients with rheumatoid arthritis treated with adalimumab, etanercept, or infliximab: results from eight years of surveillance of clinical practice in the nationwide Danish DANBIO registry. Arthritis Rheum 62:22–32
Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M et al (1988) Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 18:1797–1801
Hirota K, Hashimoto M, Yoshitomi H, Tanaka S, Nomura T, Yamaguchi T, Iwakura Y, Sakaguchi N, Sakaguchi S (2007) T cell self-reactivity forms a cytokine milieu for spontaneous development of IL-17+ Th cells that cause autoimmune arthritis. J Exp Med 204:41–47
Hoge J, Yan I, Janner N, Schumacher V, Chalaris A, Steinmetz OM, Engel DR, Scheller J, Rose-John S, Mittrucker HW (2013) IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J Immunol 190:703–711
Honorati MC, Neri S, Cattini L, Facchini A (2006) Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthr Cartil 14:345–352
Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, Ikuse T, Asano M, Iwakura Y (2000) Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med 191:313–320
Hot A, Zrioual S, Lenief V, Miossec P (2012) IL-17 and tumour necrosis factor alpha combination induces a HIF-1alpha-dependent invasive phenotype in synoviocytes. Ann Rheum Dis 71:1393–1401
Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Durez P, Tak PP, Gomez-Reino JJ, Foster CS, Kim RY, Samson CM, Falk NS, Chu DS, Callanan D, Nguyen QD, Rose K, Haider A, Di Padova F (2010) Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Sci Transl Med 2:52ra72
Huizinga TW, Fleischmann RM, Jasson M, Radin AR, van Adelsberg J, Fiore S, Huang X, Yancopoulos GD, Stahl N, Genovese MC (2014) Sarilumab, a fully human monoclonal antibody against IL-6Ralpha in patients with rheumatoid arthritis and an inadequate response to methotrexate: efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann Rheum Dis 73:1626–1634
Hunter CA, Jones SA (2015) IL-6 as a keystone cytokine in health and disease. Nat Immunol 16:448–457
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y (2009) Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30:108–119
Isomaki P, Junttila I, Vidqvist KL, Korpela M, Silvennoinen O (2015) The activity of JAK-STAT pathways in rheumatoid arthritis: constitutive activation of STAT3 correlates with interleukin 6 levels. Rheumatology (Oxford) 54:1103–1113
Iwakura Y, Ishigame H, Saijo S, Nakae S (2011) Functional specialization of interleukin-17 family members. Immunity 34:149–162
Jacques C, Gosset M, Berenbaum F, Gabay C (2006) The role of IL-1 and IL-1Ra in joint inflammation and cartilage degradation. Vitam Horm 74:371–403
Jones GW, McLoughlin RM, Hammond VJ, Parker CR, Williams JD, Malhotra R, Scheller J, Williams AS, Rose-John S, Topley N, Jones SA (2010) Loss of CD4+ T cell IL-6R expression during inflammation underlines a role for IL-6 trans signaling in the local maintenance of Th17 cells. J Immunol 184:2130–2139
Jones SA (2005) Directing transition from innate to acquired immunity: defining a role for IL-6. J Immunol 175:3463–3468
Jones SA, Scheller J, Rose-John S (2011) Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest 121:3375–3383
Joosten LA, Helsen MM, Saxne T, van De Loo FA, Heinegard D, van Den Berg WB (1999) IL-1 alpha beta blockade prevents cartilage and bone destruction in murine type II collagen-induced arthritis, whereas TNF-alpha blockade only ameliorates joint inflammation. J Immunol 163:5049–5055
Jostock T, Mullberg J, Ozbek S, Atreya R, Blinn G, Voltz N, Fischer M, Neurath MF, Rose-John S (2001) Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur J Biochem 268:160–167
Jourdan M, Bataille R, Seguin J, Zhang XG, Chaptal PA, Klein B (1990) Constitutive production of interleukin-6 and immunologic features in cardiac myxomas. Arthritis Rheum 33:398–402
Jouvenne P, Fossiez F, Banchereau J, Miossec P (1997) High levels of neutralizing autoantibodies against IL-1 alpha are associated with a better prognosis in chronic polyarthritis: a follow-up study. Scand J Immunol 46:413–418
Kalliolias GD, Ivashkiv LB (2016) TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 12:49–62
Katchamart W, Johnson S, Lin HJ, Phumethum V, Salliot C, Bombardier C (2010) Predictors for remission in rheumatoid arthritis patients: a systematic review. Arthritis Care Res (Hoboken) 62:1128–1143
Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, Kollias G (1991) Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J 10:4025–4031
Kievit W, Adang EM, Fransen J, Kuper HH, van de Laar MA, Jansen TL, De Gendt CM, De Rooij DJ, Brus HL, Van Oijen PC, Van Riel PC (2008) The effectiveness and medication costs of three anti-tumour necrosis factor alpha agents in the treatment of rheumatoid arthritis from prospective clinical practice data. Ann Rheum Dis 67:1229–1234
King IL, Dickendesher TL, Segal BM (2009) Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 113:3190–3197
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ (2006) Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two-year prospective study (the DAMAGE study cohort). Arthritis Rheum 54:1122–1131
Koenders MI, Lubberts E, Oppers-Walgreen B, van den Bersselaar L, Helsen MM, Di Padova FE, Boots AM, Gram H, Joosten LA, van den Berg WB (2005) Blocking of interleukin-17 during reactivation of experimental arthritis prevents joint inflammation and bone erosion by decreasing RANKL and interleukin-1. Am J Pathol 167:141–149
Koenders MI, van den Berg WB (2016) Secukinumab for rheumatology: development and its potential place in therapy. Drug Des Devel Ther 10:2069–2080
Kolls JK, Linden A (2004) Interleukin-17 family members and inflammation. Immunity 21:467–476
Kopf M, Baumann H, Freer G, Freudenberg M, Lamers M, Kishimoto T, Zinkernagel R, Bluethmann H, Kohler G (1994) Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368:339–342
Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL (2000) TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest 106:1481–1488
Lawlor KE, Wong PK, Campbell IK, van Rooijen N, Wicks IP (2005) Acute CD4+ T lymphocyte-dependent interleukin-1-driven arthritis selectively requires interleukin-2 and interleukin-4, joint macrophages, granulocyte-macrophage colony-stimulating factor, interleukin-6, and leukemia inhibitory factor. Arthritis Rheum 52:3749–3754
Lazzerini PE, Capecchi PL, Guidelli GM, Selvi E, Acampa M, Laghi-Pasini F (2016) Spotlight on sirukumab for the treatment of rheumatoid arthritis: the evidence to date. Drug Des Devel Ther 10:3083–3098
Lee A, Qiao Y, Grigoriev G, Chen J, Park-Min KH, Park SH, Ivashkiv LB, Kalliolias GD (2013) Tumor necrosis factor alpha induces sustained signaling and a prolonged and unremitting inflammatory response in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 65:928–938
Leizer T, Cebon J, Layton JE, Hamilton JA (1990) Cytokine regulation of colony-stimulating factor production in cultured human synovial fibroblasts: I. Induction of GM-CSF and G-CSF production by interleukin-1 and tumor necrosis factor. Blood 76:1989–1996
Liontos LM, Dissanayake D, Ohashi PS, Weiss A, Dragone LL, McGlade CJ (2011) The Src-like adaptor protein regulates GM-CSFR signaling and monocytic dendritic cell maturation. J Immunol 186:1923–1933
Listing J, Strangfeld A, Kary S, Rau R, von Hinueber U, Stoyanova-Scholz M, Gromnica-Ihle E, Antoni C, Herzer P, Kekow J, Schneider M, Zink A (2005) Infections in patients with rheumatoid arthritis treated with biologic agents. Arthritis Rheum 52:3403–3412
Liu B, Tan W, Barsoum A, Gu X, Chen K, Huang W, Ramsay A, Kolls JK, Schwarzenberger P (2010) IL-17 is a potent synergistic factor with GM-CSF in mice in stimulating myelopoiesis, dendritic cell expansion, proliferation, and functional enhancement. Exp Hematol 38(877–884):e871
Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X (2009) Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal 2:ra63
Liu F, Poursine-Laurent J, Wu HY, Link DC (1997) Interleukin-6 and the granulocyte colony-stimulating factor receptor are major independent regulators of granulopoiesis in vivo but are not required for lineage commitment or terminal differentiation. Blood 90:2583–2590
Locksley RM, Killeen N, Lenardo MJ (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104:487–501
Lopetuso LR, Chowdhry S, Pizarro TT (2013) Opposing functions of classic and novel IL-1 family members in gut health and disease. Front Immunol 4:181
Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB (2002) Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflamm Res 51:102–104
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50:650–659
Lubberts E, Schwarzenberger P, Huang W, Schurr JR, Peschon JJ, van den Berg WB, Kolls JK (2005) Requirement of IL-17 receptor signaling in radiation-resistant cells in the joint for full progression of destructive synovitis. J Immunol 175:3360–3368
Ma Y, Thornton S, Boivin GP, Hirsh D, Hirsch R, Hirsch E (1998) Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum 41:1798–1805
Madhok R, Crilly A, Watson J, Capell HA (1993) Serum interleukin 6 levels in rheumatoid arthritis: correlations with clinical and laboratory indices of disease activity. Ann Rheum Dis 52:232–234
Malemud CJ (2013) Intracellular signaling pathways in rheumatoid arthritis. J Clin Cell Immunol 4:160
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23:549–555
Martin DA, Churchill M, Flores-Suarez L, Cardiel MH, Wallace D, Martin R, Phillips K, Kaine JL, Dong H, Salinger D, Stevens E, Russell CB, Chung JB (2013) A phase Ib multiple ascending dose study evaluating safety, pharmacokinetics, and early clinical response of brodalumab, a human anti-IL-17R antibody, in methotrexate-resistant rheumatoid arthritis. Arthritis Res Ther 15:R164
McDermott MF (2009) Rilonacept in the treatment of chronic inflammatory disorders. Drugs Today (Barc) 45:423–430
McInnes IB, Buckley CD, Isaacs JD (2016) Cytokines in rheumatoid arthritis—shaping the immunological landscape. Nat Rev Rheumatol 12:63–68
McInnes IB, Schett G (2007) Cytokines in the pathogenesis of rheumatoid arthritis. Nat Rev Immunol 7:429–442
Md Yusof MY, Emery P (2013) Targeting interleukin-6 in rheumatoid arthritis. Drugs 73:341–356
Mease P, Strand V, Shalamberidze L, Dimic A, Raskina T, Xu LA, Liu Y, Smith J (2012) A phase II, double-blind, randomised, placebo-controlled study of BMS945429 (ALD518) in patients with rheumatoid arthritis with an inadequate response to methotrexate. Ann Rheum Dis 71:1183–1189
Mertens M, Singh JA (2009) Anakinra for rheumatoid arthritis: a systematic review. J Rheumatol 36:1118–1125
Metawi SA, Abbas D, Kamal MM, Ibrahim MK (2011) Serum and synovial fluid levels of interleukin-17 in correlation with disease activity in patients with RA. Clin Rheumatol 30:1201–1207
Metcalf D (2010) The colony-stimulating factors and cancer. Nat Rev Cancer 10:425–434
Metcalf D (2008) Hematopoietic cytokines. Blood 111:485–491
Mihara M, Moriya Y, Kishimoto T, Ohsugi Y (1995) Interleukin-6 (IL-6) induces the proliferation of synovial fibroblastic cells in the presence of soluble IL-6 receptor. Br J Rheumatol 34:321–325
Miller EA, Ernst JD (2008) Illuminating the black box of TNF action in tuberculous granulomas. Immunity 29:175–177
Miossec P (2002) Anti-interleukin 1alpha autoantibodies. Ann Rheum Dis 61:577–579
Miossec P (2003) Interleukin-17 in rheumatoid arthritis: if T cells were to contribute to inflammation and destruction through synergy. Arthritis Rheum 48:594–601
Miossec P, Kolls JK (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11:763–776
Miossec P, Korn T, Kuchroo VK (2009) Interleukin-17 and type 17 helper T cells. N Engl J Med 361:888–898
Moreland LW, Baumgartner SW, Schiff MH, Tindall EA, Fleischmann RM, Weaver AL, Ettlinger RE, Cohen S, Koopman WJ, Mohler K, Widmer MB, Blosch CM (1997) Treatment of rheumatoid arthritis with a recombinant human tumor necrosis factor receptor (p75)-Fc fusion protein. N Engl J Med 337:141–147
Nadkarni S, Mauri C, Ehrenstein MR (2007) Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J Exp Med 204:33–39
Nagy G, van Vollenhoven RF (2015) Sustained biologic-free and drug-free remission in rheumatoid arthritis, where are we now? Arthritis Res Ther 17:181
Nakae S, Nambu A, Sudo K, Iwakura Y (2003) Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171:6173–6177
Narazaki M, Yasukawa K, Saito T, Ohsugi Y, Fukui H, Koishihara Y, Yancopoulos GD, Taga T, Kishimoto T (1993) Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 82:1120–1126
Ndongo-Thiam N, Clement A, Pin JJ, Razanajaona-Doll D, Miossec P (2016) Negative association between autoantibodies against IL-17, IL-17/anti-IL-17 antibody immune complexes and destruction in rheumatoid arthritis. Ann Rheum Dis 75:1420–1422
Ndongo-Thiam N, Miossec P (2015) A cell-based bioassay for circulating bioactive IL-17: application to destruction in rheumatoid arthritis. Ann Rheum Dis 74:1629–1631
Netea MG, Simon A, van de Veerdonk F, Kullberg BJ, Van der Meer JW, Joosten LA (2010) IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog 6:e1000661
Neveu WA, Allard JB, Dienz O, Wargo MJ, Ciliberto G, Whittaker LA, Rincon M (2009) IL-6 is required for airway mucus production induced by inhaled fungal allergens. J Immunol 183:1732–1738
Nishimoto N, Hashimoto J, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Murata N, van der Heijde D, Kishimoto T (2007) Study of active controlled monotherapy used for rheumatoid arthritis, an IL-6 inhibitor (SAMURAI): evidence of clinical and radiographic benefit from an X ray reader-blinded randomised controlled trial of tocilizumab. Ann Rheum Dis 66:1162–1167
Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Hashimoto J, Azuma J, Kishimoto T (2004) Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum 50:1761–1769
Noack M, Ndongo-Thiam N, Miossec P (2016) Interaction among activated lymphocytes and mesenchymal cells through podoplanin is critical for a high IL-17 secretion. Arthritis Res Ther 18:148
Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F (2003) The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci 28:226–229
O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A (2015) The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66:311–328
Ohshima S, Saeki Y, Mima T, Sasai M, Nishioka K, Nomura S, Kopf M, Katada Y, Tanaka T, Suemura M, Kishimoto T (1998) Interleukin 6 plays a key role in the development of antigen-induced arthritis. Proc Natl Acad Sci U S A 95:8222–8226
Osta B, Benedetti G, Miossec P (2014) Classical and paradoxical effects of TNF-alpha on bone homeostasis. Front Immunol 5:48
Osta B, Lavocat F, Eljaafari A, Miossec P (2014) Effects of interleukin-17A on osteogenic differentiation of isolated human mesenchymal stem cells. Front Immunol 5:425
Paleolog EM (2002) Angiogenesis in rheumatoid arthritis. Arthritis Res 4(Suppl 3):S81–S90
Pavelka K, Chon Y, Newmark R, Lin SL, Baumgartner S, Erondu N (2015) A study to evaluate the safety, tolerability, and efficacy of brodalumab in subjects with rheumatoid arthritis and an inadequate response to methotrexate. J Rheumatol 42:912–919
Pereira J, Velloso ED, Loterio HA, Laurindo IM, Chamone DA (1994) Long-term remission of neutropenia in Felty’s syndrome after a short GM-CSF treatment. Acta Haematol 92:154–156
Peters M, Jacobs S, Ehlers M, Vollmer P, Mullberg J, Wolf E, Brem G, KH M z B, Rose-John S (1996) The function of the soluble interleukin 6 (IL-6) receptor in vivo: sensitization of human soluble IL-6 receptor transgenic mice towards IL-6 and prolongation of the plasma half-life of IL-6. J Exp Med 183:1399–1406
Pettipher ER, Higgs GA, Henderson B (1986) Interleukin 1 induces leukocyte infiltration and cartilage proteoglycan degradation in the synovial joint. Proc Natl Acad Sci U S A 83:8749–8753
Plater-Zyberk C, Joosten LA, Helsen MM, Hepp J, Baeuerle PA, van den Berg WB (2007) GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Ann Rheum Dis 66:452–457
Plater-Zyberk C, Joosten LA, Helsen MM, Koenders MI, Baeuerle PA, van den Berg WB (2009) Combined blockade of granulocyte-macrophage colony stimulating factor and interleukin 17 pathways potently suppresses chronic destructive arthritis in a tumour necrosis factor alpha-independent mouse model. Ann Rheum Dis 68:721–728
Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN (2007) GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 178:39–48
Probert L (2015) TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 302:2–22
Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X (2007) The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8:247–256
Ramiro S, Gaujoux-Viala C, Nam JL, Smolen JS, Buch M, Gossec L, van der Heijde D, Winthrop K, Landewe R (2014) Safety of synthetic and biological DMARDs: a systematic literature review informing the 2013 update of the EULAR recommendations for management of rheumatoid arthritis. Ann Rheum Dis 73:529–535
Rose-John S, Neurath MF (2004) IL-6 trans-signaling: the heat is on. Immunity 20:2–4
Sack U, Kinne RW, Marx T, Heppt P, Bender S, Emmrich F (1993) Interleukin-6 in synovial fluid is closely associated with chronic synovitis in rheumatoid arthritis. Rheumatol Int 13:45–51
Schett G, Dayer JM, Manger B (2015) Interleukin-1 function and role in rheumatic disease. Nat Rev Rheumatol 12:14–24
Schett G, Elewaut D, McInnes IB, Dayer JM, Neurath MF (2013) How cytokine networks fuel inflammation: toward a cytokine-based disease taxonomy. Nat Med 19:822–824
Schweizerhof M, Stosser S, Kurejova M, Njoo C, Gangadharan V, Agarwal N, Schmelz M, Bali KK, Michalski CW, Brugger S, Dickenson A, Simone DA, Kuner R (2009) Hematopoietic colony-stimulating factors mediate tumor-nerve interactions and bone cancer pain. Nat Med 15:802–807
Sesin CA, Bingham CO 3rd (2005) Remission in rheumatoid arthritis: wishful thinking or clinical reality? Semin Arthritis Rheum 35:185–196
Shetty A, Hanson R, Korsten P, Shawagfeh M, Arami S, Volkov S, Vila O, Swedler W, Shunaigat AN, Smadi S, Sawaqed R, Perkins D, Shahrara S, Sweiss NJ (2014) Tocilizumab in the treatment of rheumatoid arthritis and beyond. Drug Des Devel Ther 8:349–364
Shibata Y, Berclaz PY, Chroneos ZC, Yoshida M, Whitsett JA, Trapnell BC (2001) GM-CSF regulates alveolar macrophage differentiation and innate immunity in the lung through PU.1. Immunity 15:557–567
Shiomi A, Usui T (2015) Pivotal roles of GM-CSF in autoimmunity and inflammation. Mediat Inflamm 2015:568543
Shiomi A, Usui T, Ishikawa Y, Shimizu M, Murakami K, Mimori T (2014) GM-CSF but not IL-17 is critical for the development of severe interstitial lung disease in SKG mice. J Immunol 193:849–859
Sims JE, Smith DE (2010) The IL-1 family: regulators of immunity. Nat Rev Immunol 10:89–102
Singh JA, Furst DE, Bharat A, Curtis JR, Kavanaugh AF, Kremer JM, Moreland LW, O’Dell J, Winthrop KL, Beukelman T, Bridges SL Jr, Chatham WW, Paulus HE, Suarez-Almazor M, Bombardier C, Dougados M, Khanna D, King CM, Leong AL, Matteson EL, Schousboe JT, Moynihan E, Kolba KS, Jain A, Volkmann ER, Agrawal H, Bae S, Mudano AS, Patkar NM, Saag KG (2012) 2012 update of the 2008 American College of Rheumatology recommendations for the use of disease-modifying antirheumatic drugs and biologic agents in the treatment of rheumatoid arthritis. Arthritis Care Res (Hoboken) 64:625–639
Skiniotis G, Boulanger MJ, Garcia KC, Walz T (2005) Signaling conformations of the tall cytokine receptor gp130 when in complex with IL-6 and IL-6 receptor. Nat Struct Mol Biol 12:545–551
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis. Lancet 388:2023–2038
Smolen JS, Beaulieu A, Rubbert-Roth A, Ramos-Remus C, Rovensky J, Alecock E, Woodworth T, Alten R (2008) Effect of interleukin-6 receptor inhibition with tocilizumab in patients with rheumatoid arthritis (OPTION study): a double-blind, placebo-controlled, randomised trial. Lancet 371:987–997
Smolen JS, Landewe R, Breedveld FC, Dougados M, Emery P, Gaujoux-Viala C, Gorter S, Knevel R, Nam J, Schoels M, Aletaha D, Buch M, Gossec L, Huizinga T, Bijlsma JW, Burmester G, Combe B, Cutolo M, Gabay C, Gomez-Reino J, Kouloumas M, Kvien TK, Martin-Mola E, McInnes I, Pavelka K, van Riel P, Scholte M, Scott DL, Sokka T, Valesini G, van Vollenhoven R, Winthrop KL, Wong J, Zink A, van der Heijde D (2010) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs. Ann Rheum Dis 69:964–975
Smolen JS, Weinblatt ME, Sheng S, Zhuang Y, Hsu B (2014) Sirukumab, a human anti-interleukin-6 monoclonal antibody: a randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 73:1616–1625
Stojanov S, Kastner DL (2005) Familial autoinflammatory diseases: genetics, pathogenesis and treatment. Curr Opin Rheumatol 17:586–599
Stosser S, Schweizerhof M, Kuner R (2011) Hematopoietic colony-stimulating factors: new players in tumor-nerve interactions. J Mol Med (Berl) 89:321–329
Sugarman BJ, Aggarwal BB, Hass PE, Figari IS, Palladino MA Jr, Shepard HM (1985) Recombinant human tumor necrosis factor-alpha: effects on proliferation of normal and transformed cells in vitro. Science 230:943–945
Suzuki M, Hashizume M, Yoshida H, Mihara M (2010) Anti-inflammatory mechanism of tocilizumab, a humanized anti-IL-6R antibody: effect on the expression of chemokine and adhesion molecule. Rheumatol Int 30:309–315
Suzuki M, Hashizume M, Yoshida H, Shiina M, Mihara M (2010) IL-6 and IL-1 synergistically enhanced the production of MMPs from synovial cells by up-regulating IL-6 production and IL-1 receptor I expression. Cytokine 51:178–183
Tanaka Y, Martin Mola E (2014) IL-6 targeting compared to TNF targeting in rheumatoid arthritis: studies of olokizumab, sarilumab and sirukumab. Ann Rheum Dis 73:1595–1597
Tarnowski M, Paradowska-Gorycka A, Dabrowska-Zamojcin E, Czerewaty M, Sluczanowska-Glabowska S, Pawlik A (2016) The effect of gene polymorphisms on patient responses to rheumatoid arthritis therapy. Expert Opin Drug Metab Toxicol 12:41–55
Taylor PC, Peters AM, Paleolog E, Chapman PT, Elliott MJ, McCloskey R, Feldmann M, Maini RN (2000) Reduction of chemokine levels and leukocyte traffic to joints by tumor necrosis factor alpha blockade in patients with rheumatoid arthritis. Arthritis Rheum 43:38–47
Thompson C, Davies R, Choy E (2016) Anti cytokine therapy in chronic inflammatory arthritis. Cytokine 86:92–99
Toh ML, Gonzales G, Koenders MI, Tournadre A, Boyle D, Lubberts E, Zhou Y, Firestein GS, van den Berg WB, Miossec P (2010) Role of interleukin 17 in arthritis chronicity through survival of synoviocytes via regulation of synoviolin expression. PLoS One 5:e13416
Toh ML, Kawashima M, Hot A, Miossec P (2010) Role of IL-17 in the Th1 systemic defects in rheumatoid arthritis through selective IL-12Rbeta2 inhibition. Ann Rheum Dis 69:1562–1567
Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J (2006) Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 177:36–39
Uchiyama Y, Yorozu K, Hashizume M, Moriya Y, Mihara M (2008) Tocilizumab, a humanized anti-interleukin-6 receptor antibody, ameliorates joint swelling in established monkey collagen-induced arthritis. Biol Pharm Bull 31:1159–1163
Ulfgren AK, Grondal L, Lindblad S, Khademi M, Johnell O, Klareskog L, Andersson U (2000) Interindividual and intra-articular variation of proinflammatory cytokines in patients with rheumatoid arthritis: potential implications for treatment. Ann Rheum Dis 59:439–447
van de Loo FA, Joosten LA, van Lent PL, Arntz OJ, van den Berg WB (1995) Role of interleukin-1, tumor necrosis factor alpha, and interleukin-6 in cartilage proteoglycan metabolism and destruction. Effect of in situ blocking in murine antigen- and zymosan-induced arthritis. Arthritis Rheum 38:164–172
van der Meer JW, Barza M, Wolff SM, Dinarello CA (1988) A low dose of recombinant interleukin 1 protects granulocytopenic mice from lethal gram-negative infection. Proc Natl Acad Sci U S A 85:1620–1623
Van Lent PL, Van De Loo FA, Holthuysen AE, Van Den Bersselaar LA, Vermeer H, Van Den Berg WB (1995) Major role for interleukin 1 but not for tumor necrosis factor in early cartilage damage in immune complex arthritis in mice. J Rheumatol 22:2250–2258
van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP (2013) GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol 56:675–682
van Nieuwenhuijze AE, van de Loo FA, Walgreen B, Bennink M, Helsen M, van den Bersselaar L, Wicks IP, van den Berg WB, Koenders MI (2015) Complementary action of granulocyte macrophage colony-stimulating factor and interleukin-17A induces interleukin-23, receptor activator of nuclear factor-kappaB ligand, and matrix metalloproteinases and drives bone and cartilage pathology in experimental arthritis: rationale for combination therapy in rheumatoid arthritis. Arthritis Res Ther 17:163
Wallis RS (2008) Tumour necrosis factor antagonists: structure, function, and tuberculosis risks. Lancet Infect Dis 8:601–611
Weinblatt ME, Mease P, Mysler E, Takeuchi T, Drescher E, Berman A, Xing J, Zilberstein M, Banerjee S, Emery P (2015) The efficacy and safety of subcutaneous clazakizumab in patients with moderate-to-severe rheumatoid arthritis and an inadequate response to methotrexate: results from a multinational, phase IIb, randomized, double-blind, placebo/active-controlled, dose-ranging study. Arthritis Rheumatol 67:2591–2600
Wicks IP, Roberts AW (2016) Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol 12:37–48
Williams RO, Feldmann M, Maini RN (1992) Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A 89:9784–9788
Wooley PH, Dutcher J, Widmer MB, Gillis S (1993) Influence of a recombinant human soluble tumor necrosis factor receptor FC fusion protein on type II collagen-induced arthritis in mice. J Immunol 151:6602–6607
Xu WD, Firestein GS, Taetle R, Kaushansky K, Zvaifler NJ (1989) Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J Clin Invest 83:876–882
Yang YH, Hamilton JA (2001) Dependence of interleukin-1-induced arthritis on granulocyte-macrophage colony-stimulating factor. Arthritis Rheum 44:111–119
Yoshizaki K, Matsuda T, Nishimoto N, Kuritani T, Taeho L, Aozasa K, Nakahata T, Kawai H, Tagoh H, Komori T et al (1989) Pathogenic significance of interleukin-6 (IL-6/BSF-2) in Castleman’s disease. Blood 74:1360–1367
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W (2000) High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol 164:2832–2838
Zrioual S, Toh ML, Tournadre A, Zhou Y, Cazalis MA, Pachot A, Miossec V, Miossec P (2008) IL-17RA and IL-17RC receptors are essential for IL-17A-induced ELR+ CXC chemokine expression in synoviocytes and are overexpressed in rheumatoid blood. J Immunol 180:655–663
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Additional information
This article is a contribution to the special issue on Immunopathology of Rheumatoid Arthritis - Guest Editors: Cem Gabay and Paul Hasler
Rights and permissions
About this article

Cite this article
Noack, M., Miossec, P. Selected cytokine pathways in rheumatoid arthritis. Semin Immunopathol 39, 365–383 (2017). https://doi.org/10.1007/s00281-017-0619-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-017-0619-z